
Page 60 - Read Online
P. 60
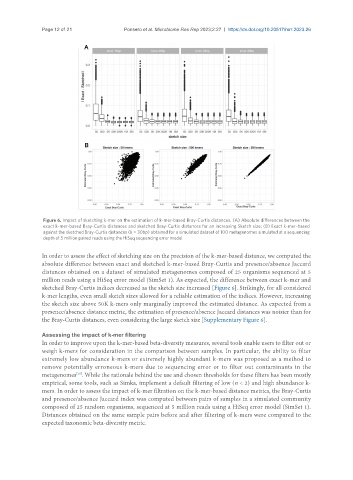
Page 12 of 21 Ponsero et al. Microbiome Res Rep 2023;2:27 https://dx.doi.org/10.20517/mrr.2023.26
Figure 6. Impact of sketching k-mer on the estimation of k-mer-based Bray-Curtis distances. (A) Absolute differences between the
exact k-mer-based Bray-Curtis distances and sketched Bray-Curtis distances for an increasing Sketch size; (B) Exact k-mer-based
against the sketched Bray-Curtis distances (k = 30bp) obtained for a simulated dataset of 100 metagenomes simulated at a sequencing
depth of 5 million paired reads using the HiSeq sequencing error model.
In order to assess the effect of sketching size on the precision of the k-mer-based distance, we computed the
absolute difference between exact and sketched k-mer-based Bray-Curtis and presence/absence Jaccard
distances obtained on a dataset of simulated metagenomes composed of 25 organisms sequenced at 5
million reads using a HiSeq error model (SimSet 1). As expected, the difference between exact k-mer and
sketched Bray-Curtis indices decreased as the sketch size increased [Figure 6]. Strikingly, for all considered
k-mer lengths, even small sketch sizes allowed for a reliable estimation of the indices. However, increasing
the sketch size above 50K k-mers only marginally improved the estimated distance. As expected from a
presence/absence distance metric, the estimation of presence/absence Jaccard distances was noisier than for
the Bray-Curtis distances, even considering the large sketch size [Supplementary Figure 6].
Assessing the impact of k-mer filtering
In order to improve upon the k-mer-based beta-diversity measures, several tools enable users to filter out or
weigh k-mers for consideration in the comparison between samples. In particular, the ability to filter
extremely low abundance k-mers or extremely highly abundant k-mers was proposed as a method to
remove potentially erroneous k-mers due to sequencing error or to filter out contaminants in the
metagenomes . While the rationale behind the use and chosen thresholds for these filters has been mostly
[10]
empirical, some tools, such as Simka, implement a default filtering of low (n < 2) and high abundance k-
mers. In order to assess the impact of k-mer filtration on the k-mer-based distance metrics, the Bray-Curtis
and presence/absence Jaccard index was computed between pairs of samples in a simulated community
composed of 25 random organisms, sequenced at 5 million reads using a HiSeq error model (SimSet 1).
Distances obtained on the same sample pairs before and after filtering of k-mers were compared to the
expected taxonomic beta-diversity metric.