
Page 56 - Read Online
P. 56
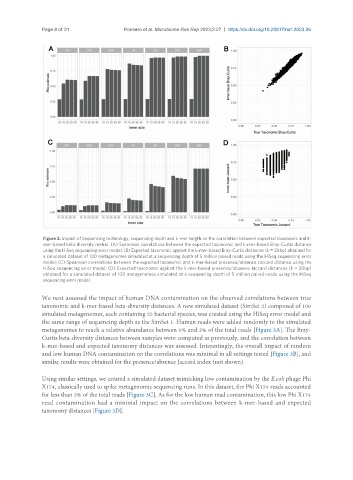
Page 8 of 21 Ponsero et al. Microbiome Res Rep 2023;2:27 https://dx.doi.org/10.20517/mrr.2023.26
Figure 2. Impact of Sequencing technology, sequencing depth and k-mer length on the correlation between expected taxonomic and k-
mer-based beta-diversity metric. (A) Spearman correlations between the expected taxonomic and k-mer-based Bray-Curtis distance
using the HiSeq sequencing error model; (B) Expected taxonomic against the k-mer-based Bray-Curtis distances (k = 30bp) obtained for
a simulated dataset of 100 metagenomes simulated at a sequencing depth of 5 million paired reads using the HiSeq sequencing error
model; (C) Spearman correlations between the expected taxonomic and k-mer-based presence/absence Jaccard distance using the
HiSeq sequencing error model; (D) Expected taxonomic against the k-mer-based presence/absence Jaccard distances (k = 30bp)
obtained for a simulated dataset of 100 metagenomes simulated at a sequencing depth of 5 million paired-reads using the HiSeq
sequencing error model.
We next assessed the impact of human DNA contamination on the observed correlations between true
taxonomic and k-mer-based beta-diversity distances. A new simulated dataset (SimSet 2) composed of 100
simulated metagenomes, each containing 25 bacterial species, was created using the HiSeq error model and
the same range of sequencing depth as the SimSet 1. Human reads were added randomly to the simulated
metagenomes to reach a relative abundance between 0% and 2% of the total reads [Figure 3A]. The Bray-
Curtis beta-diversity distances between samples were computed as previously, and the correlation between
k-mer-based and expected taxonomy distances was assessed. Interestingly, the overall impact of random
and low human DNA contamination on the correlations was minimal in all settings tested [Figure 3B], and
similar results were obtained for the presence/absence Jaccard index (not shown).
Using similar settings, we created a simulated dataset mimicking low contamination by the E.coli phage Phi
X174, classically used to spike metagenomic sequencing runs. In this dataset, the Phi X174 reads accounted
for less than 2% of the total reads [Figure 3C]. As for the low human read contamination, this low Phi X174
read contamination had a minimal impact on the correlations between k-mer-based and expected
taxonomy distances [Figure 3D].