
Page 246 - Read Online
P. 246
Page 8 of 10 Raevsky et al. Neuroimmunol Neuroinflammation 2018;5:33 I http://dx.doi.org/10.20517/2347-8659.2018.34
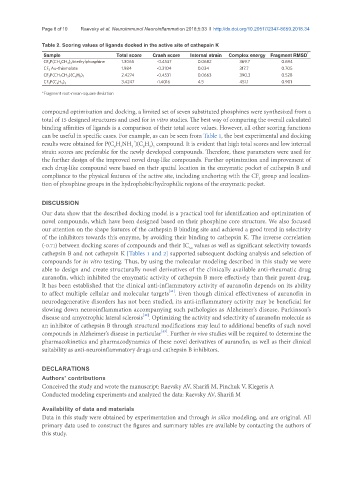
Table 2. Scoring values of ligands docked in the active site of cathepsin K
Sample Total score Crash score Internal strain Complex energy Fragment RMSD *
CF 3 P(CH 2 CH 3 ) 3 triethylphosphine 1.3065 -0.4537 0.0682 369.7 0.694
CF 3 Au-thiomalate 1.984 -0.3104 0.034 317.7 0.705
2.4274 -0.4531 0.0663 390.3 0.528
CF 3 P(CH 2 CH 3 )(C 6 H 5 ) 2
3.4247 -1.4016 4.5 451.1 0.901
CF 3 P(C 6 H 5 ) 3
*Fragment root-mean-square deviation
compound optimization and docking, a limited set of seven substituted phosphines were synthesized from a
total of 15 designed structures and used for in vitro studies. The best way of comparing the overall calculated
binding affinities of ligands is a comparison of their total score values. However, all other scoring functions
can be useful in specific cases. For example, as can be seen from Table 1, the best experimental and docking
results were obtained for P(C H NH )(C H ) compound. It is evident that high total scores and low internal
+
6
3
6
5 2
4
strain scores are preferable for the newly developed compounds. Therefore, these parameters were used for
the further design of the improved novel drug-like compounds. Further optimization and improvement of
each drug-like compound were based on their spatial location in the enzymatic pocket of cathepsin B and
compliance to the physical features of the active site, including anchoring with the CF group and localiza-
3
tion of phosphine groups in the hydrophobic/hydrophilic regions of the enzymatic pocket.
DISCUSSION
Our data show that the described docking model is a practical tool for identification and optimization of
novel compounds, which have been designed based on their phosphine core structure. We also focused
our attention on the shape features of the cathepsin B binding site and achieved a good trend in selectivity
of the inhibitors towards this enzyme, by avoiding their binding to cathepsin K. The inverse correlation
(-0.71) between docking scores of compounds and their IC values as well as significant selectivity towards
50
cathepsin B and not cathepsin K [Tables 1 and 2] supported subsequent docking analysis and selection of
compounds for in vitro testing. Thus, by using the molecular modeling described in this study we were
able to design and create structurally novel derivatives of the clinically available anti-rheumatic drug
auranofin, which inhibited the enzymatic activity of cathepsin B more effectively than their parent drug.
It has been established that the clinical anti-inflammatory activity of auranofin depends on its ability
to affect multiple cellular and molecular targets . Even though clinical effectiveness of auranofin in
[41]
neurodegenerative disorders has not been studied, its anti-inflammatory activity may be beneficial for
slowing down neuroinflammation accompanying such pathologies as Alzheimer’s disease, Parkinson’s
[42]
disease and amyotrophic lateral sclerosis . Optimizing the activity and selectivity of auranofin molecule as
an inhibitor of cathepsin B through structural modifications may lead to additional benefits of such novel
[43]
compounds in Alzheimer’s disease in particular . Further in vivo studies will be required to determine the
pharmacokinetics and pharmacodynamics of these novel derivatives of auranofin, as well as their clinical
suitability as anti-neuroinflammatory drugs and cathepsin B inhibitors.
DECLARATIONS
Authors’ contributions
Conceived the study and wrote the manuscript: Raevsky AV, Sharifi M, Pinchuk V, Klegeris A
Conducted modeling experiments and analyzed the data: Raevsky AV, Sharifi M
Availability of data and materials
Data in this study were obtained by experimentation and through in silico modeling, and are original. All
primary data used to construct the figures and summary tables are available by contacting the authors of
this study.