
Page 80 - Read Online
P. 80
Goodman et al. J Transl Genet Genom 2020;4:144-58 I http://dx.doi.org/10.20517/jtgg.2020.23 Page 149
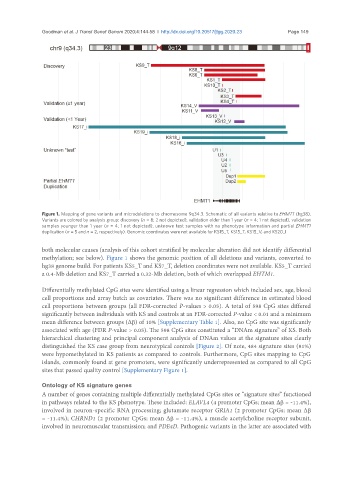
Figure 1. Mapping of gene variants and microdeletions to chromosome 9q34.3. Schematic of all variants relative to EHMT1 (hg38).
Variants are colored by analysis group: discovery (n = 8; 2 not depicted), validation older than 1 year (n = 4; 1 not depicted), validation
samples younger than 1 year (n = 4; 1 not depicted), unknown test samples with no phenotype information and partial EHMT1
duplication (n = 5 and n = 2, respectively). Genomic coordinates were not available for KS15_T, KS15_T, KS15_V, and KS20_I
both molecular causes (analysis of this cohort stratified by molecular alteration did not identify differential
methylation; see below). Figure 1 shows the genomic position of all deletions and variants, converted to
hg38 genome build. For patients KS5_T and KS7_T, deletion coordinates were not available. KS5_T carried
a 0.4-Mb deletion and KS7_T carried a 0.32-Mb deletion, both of which overlapped EHTM1.
Differentially methylated CpG sites were identified using a linear regression which included sex, age, blood
cell proportions and array batch as covariates. There was no significant difference in estimated blood
cell proportions between groups (all FDR-corrected P-values > 0.05). A total of 598 CpG sites differed
significantly between individuals with KS and controls at an FDR-corrected P-value < 0.01 and a minimum
mean difference between groups (Δb) of 10% [Supplementary Table 1]. Also, no CpG site was significantly
associated with age (FDR P-value > 0.05). The 598 CpG sites constituted a “DNAm signature” of KS. Both
hierarchical clustering and principal component analysis of DNAm values at the signature sites clearly
distinguished the KS case group from neurotypical controls [Figure 2]. Of note, 484 signature sites (81%)
were hypomethylated in KS patients as compared to controls. Furthermore, CpG sites mapping to CpG
islands, commonly found at gene promoters, were significantly underrepresented as compared to all CpG
sites that passed quality control [Supplementary Figure 1].
Ontology of KS signature genes
A number of genes containing multiple differentially methylated CpGs sites or “signature sites” functioned
in pathways related to the KS phenotype. These included: ELAVL4 (4 promoter CpGs; mean Δb = -11.4%),
involved in neuron-specific RNA processing; glutamate receptor GRIA1 (2 promoter CpGs; mean Δb
= -11.4%); CHRND1 (2 promoter CpGs; mean Δb = -11.4%), a muscle acetylcholine receptor subunit,
involved in neuromuscular transmission; and PDE4D. Pathogenic variants in the latter are associated with