
Page 84 - Read Online
P. 84
Goodman et al. J Transl Genet Genom 2020;4:144-58 I http://dx.doi.org/10.20517/jtgg.2020.23 Page 153
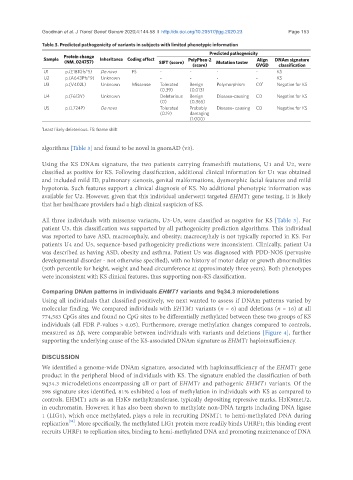
Table 3. Predicted pathogenicity of variants in subjects with limited phenotypic information
Predicted pathogenicity
Protein change
Sample Inheritance Coding effect PolyPhen-2 Align DNAm signature
(NM_024757) SIFT (score) Mutation taster
(score) GVGD classification
U1 p.(E181Gfs*5) De novo FS - - - - KS
U2 p.(A643Pfs*9) Unknown - - - - KS
U3 p.(V402L) Unknown Missense Tolerated Benign Polymorphism C0 1 Negative for KS
(0.39) (0.013)
U4 p.(F613Y) Unknown Deleterious Benign Disease-causing C0 Negative for KS
(0) (0.365)
U5 p.(L724P) De novo Tolerated Probably Disease- causing C0 Negative for KS
(0.19) damaging
(1.000)
1 Least likely deleterious. FS: frame shift
algorithms [Table 3] and found to be novel in gnomAD (v3).
Using the KS DNAm signature, the two patients carrying frameshift mutations, U1 and U2, were
classified as positive for KS. Following classification, additional clinical information for U1 was obtained
and included mild ID, pulmonary stenosis, genital malformations, dysmorphic facial features and mild
hypotonia. Such features support a clinical diagnosis of KS. No additional phenotypic information was
available for U2. However, given that this individual underwent targeted EHMT1 gene testing, it is likely
that her healthcare providers had a high clinical suspicion of KS.
All three individuals with missense variants, U3-U5, were classified as negative for KS [Table 3]. For
patient U3, this classification was supported by all pathogenicity prediction algorithms. This individual
was reported to have ASD, macrocephaly, and obesity; macrocephaly is not typically reported in KS. For
patients U4 and U5, sequence-based pathogenicity predictions were inconsistent. Clinically, patient U4
was described as having ASD, obesity and asthma. Patient U5 was diagnosed with PDD-NOS (pervasive
developmental disorder - not otherwise specified), with no history of motor delay or growth abnormalities
(50th percentile for height, weight and head circumference at approximately three years). Both phenotypes
were inconsistent with KS clinical features, thus supporting non-KS classification.
Comparing DNAm patterns in individuals EHMT1 variants and 9q34.3 microdeletions
Using all individuals that classified positively, we next wanted to assess if DNAm patterns varied by
molecular finding. We compared individuals with EHTM1 variants (n = 6) and deletions (n = 16) at all
774,583 CpGs sites and found no CpG sites to be differentially methylated between these two groups of KS
individuals (all FDR P-values > 0.05). Furthermore, average methylation changes compared to controls,
measured as Δb, were comparable between individuals with variants and deletions [Figure 4], further
supporting the underlying cause of the KS-associated DNAm signature as EHMT1 haploinsufficiency.
DISCUSSION
We identified a genome-wide DNAm signature, associated with haploinsufficiency of the EHMT1 gene
product in the peripheral blood of individuals with KS. The signature enabled the classification of both
9q34.3 microdeletions encompassing all or part of EHMT1 and pathogenic EHMT1 variants. Of the
598 signature sites identified, 81% exhibited a loss of methylation in individuals with KS as compared to
controls. EHMT1 acts as an H3K9 methyltransferase, typically depositing repressive marks, H3K9me1/2,
in euchromatin. However, it has also been shown to methylate non-DNA targets including DNA ligase
1 (LIG1), which once methylated, plays a role in recruiting DNMT1 to hemi-methylated DNA during
[38]
replication . More specifically, the methylated LIG1 protein more readily binds UHRF1; this binding event
recruits UHRF1 to replication sites, binding to hemi-methylated DNA and promoting maintenance of DNA