
Page 24 - Read Online
P. 24
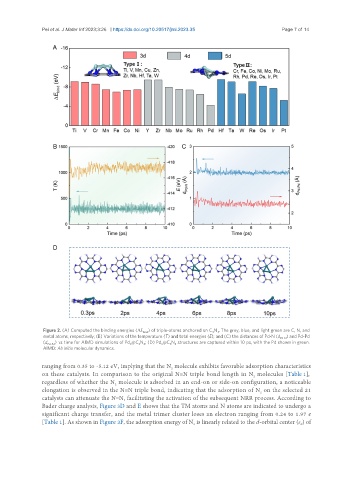
Pei et al. J Mater Inf 2023;3:26 https://dx.doi.org/10.20517/jmi.2023.35 Page 7 of 14
Figure 2. (A) Computed the binding energies (ΔE ) of triple-atoms anchored on C N . The grey, blue, and light green are C, N, and
bind 3 3
metal atoms, respectively; (B) Variations of the temperature (T) and total energies (E); and (C) the distances of Pd-N (d Pd-N ) and Pd-Pd
(d Pd-Pd ) vs. time for AIMD simulations of Pd @C N ; (D) Pd @C N structures are captured within 10 ps, with the Pd shown in green.
3
3
3
3
3
3
AIMD: Ab initio molecular dynamics.
ranging from 0.35 to -5.12 eV, implying that the N molecule exhibits favorable adsorption characteristics
2
on these catalysts. In comparison to the original N≡N triple bond length in N molecules [Table 1],
2
regardless of whether the N molecule is adsorbed in an end-on or side-on configuration, a noticeable
2
elongation is observed in the N≡N triple bond, indicating that the adsorption of N on the selected 21
2
catalysts can attenuate the N≡N, facilitating the activation of the subsequent NRR process. According to
Bader charge analysis, Figure 3D and E shows that the TM atoms and N atoms are indicated to undergo a
significant charge transfer, and the metal trimer cluster loses an electron ranging from 0.24 to 1.97 e
[Table 1]. As shown in Figure 3F, the adsorption energy of N is linearly related to the d-orbital center (ε ) of
d
2