
Page 15 - Read Online
P. 15
Page 10 of 20 Hamawandi et al. Energy Mater. 2025, 5, 500065 https://dx.doi.org/10.20517/energymater.2024.204
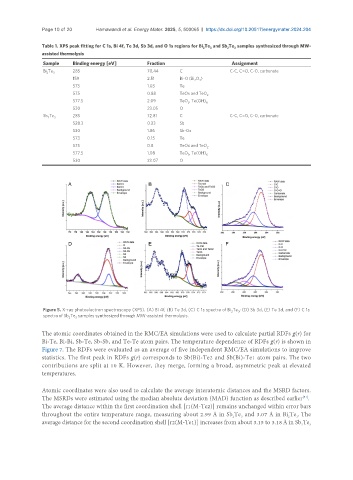
Table 1. XPS peak fitting for C 1s, Bi 4f, Te 3d, Sb 3d, and O 1s regions for Bi Te and Sb Te samples synthesized through MW-
3
2
2
3
assisted thermolysis
Sample Binding energy [eV] Fraction Assignment
Bi Te 3 285 70.44 C C-C, C=O, C-O, carbonate
2
159 2.51 Bi-O (Bi O )
2 3
573 1.03 Te
575 0.88 TeOx and TeO 2
577.5 2.09 TeO , Te(OH) 6
3
530 23.05 O
Sb Te 285 72.81 C C-C, C=O, C-O, carbonate
2 3
528.3 0.23 Sb
530 1.86 Sb-Ox
573 0.15 Te
575 0.8 TeOx and TeO
2
577.5 1.08 TeO , Te(OH) 6
3
530 23.07 O
Figure 5. X-ray photoelectron spectroscopy (XPS). (A) Bi 4f, (B) Te 3d, (C) C 1s spectra of Bi Te ; (D) Sb 3d, (E) Te 3d, and (F) C 1s
2
3
spectra of Sb Te samples synthesized through MW-assisted thermolysis.
3
2
The atomic coordinates obtained in the RMC/EA simulations were used to calculate partial RDFs g(r) for
Bi-Te, Bi-Bi, Sb-Te, Sb-Sb, and Te-Te atom pairs. The temperature dependence of RDFs g(r) is shown in
Figure 7. The RDFs were evaluated as an average of five independent RMC/EA simulations to improve
statistics. The first peak in RDFs g(r) corresponds to Sb(Bi)-Te2 and Sb(Bi)-Te1 atom pairs. The two
contributions are split at 10 K. However, they merge, forming a broad, asymmetric peak at elevated
temperatures.
Atomic coordinates were also used to calculate the average interatomic distances and the MSRD factors.
The MSRDs were estimated using the median absolute deviation (MAD) function as described earlier .
[51]
The average distance within the first coordination shell [r1(M-Te2)] remains unchanged within error bars
throughout the entire temperature range, measuring about 2.99 Å in Sb Te and 3.07 Å in Bi Te . The
2
3
2
3
average distance for the second coordination shell [r2(M-Te1)] increases from about 3.15 to 3.18 Å in Sb Te
3
2