
Page 56 - Read Online
P. 56
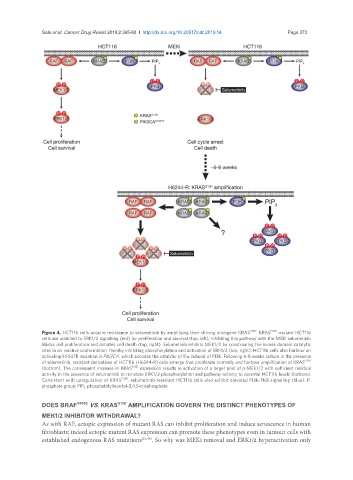
Sale et al. Cancer Drug Resist 2019;2:365-80 I http://dx.doi.org/10.20517/cdr.2019.14 Page 373
Figure 4. HCT116 cells acquire resistance to selumetinib by amplifying their driving oncogene KRAS G13D . KRAS G13D -mutant HCT116
cells are addicted to ERK1/2 signalling (red) for proliferation and survival (top, left); inhibiting this pathway with the MEKi selumetinib
blocks cell proliferation and initiates cell death (top, right). Selumetinib inhibits MEK1/2 by constraining the kinase domain catalytic
sites in an inactive conformation, thereby inhibiting phosphorylation and activation of ERK1/2 (top, right). HCT116 cells also harbour an
activating H1047R mutation in PIK3CA, which encodes the catalytic p110α subunit of PI3K. Following 6-8 weeks culture in the presence
of selumetinib, resistant derivatives of HCT116 (H6244-R) cells emerge that proliferate normally and harbour amplification of KRAS G13D
(bottom). The consequent increase in KRAS G13D expression results in activation of a larger pool of p-MEK1/2 with sufficient residual
activity in the presence of selumetinib to reinstate ERK1/2 phosphorylation and pathway activity to parental HCT116 levels (bottom).
Consistent with upregulation of KRAS G13D , selumetinib-resistant HCT116 cells also exhibit elevated PI3K-PKB signalling (blue). P:
phosphate group; PIP 3 : phosphatidylinositol-3,4,5-trisphosphate
DOES BRAF V600E VS. KRAS G13D AMPLIFICATION GOVERN THE DISTINCT PHENOTYPES OF
MEK1/2 INHIBITOR WITHDRAWAL?
As with RAF, ectopic expression of mutant RAS can inhibit proliferation and induce senescence in human
fibroblasts; indeed ectopic mutant RAS expression can promote these phenotypes even in tumour cells with
established endogenous RAS mutations [21,40] . So why was MEKi removal and ERK1/2 hyperactivation only