
Page 53 - Read Online
P. 53
Page 370 Sale et al. Cancer Drug Resist 2019;2:365-80 I http://dx.doi.org/10.20517/cdr.2019.14
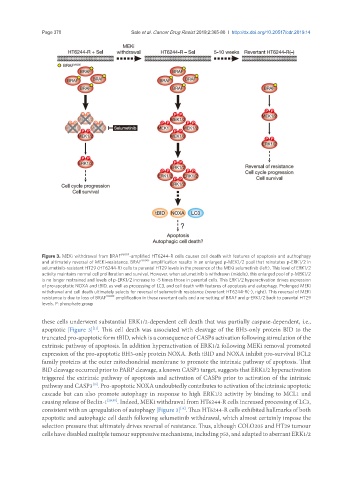
Figure 3. MEKi withdrawal from BRAF V600E -amplified HT6244-R cells causes cell death with features of apoptosis and authophagy
and ultimately reversal of MEKi-resistance. BRAF V600E amplification results in an enlarged p-MEK1/2 pool that reinstates p-ERK1/2 in
selumetinib-resistant HT29 (HT6244-R) cells to parental HT29 levels in the presence of the MEKi selumetinib (left). This level of ERK1/2
activity maintains normal cell proliferation and survival. However, when selumetinib is withdrawn (middle), this enlarged pool of p-MEK1/2
is no longer restrained and levels of p-ERK1/2 increase to ~5 times those in parental cells. This ERK1/2 hyperactivation drives expression
of pro-apoptotic NOXA and tBID, as well as processing of LC3, and cell death with features of apoptosis and autophagy. Prolonged MEKi
withdrawal and cell death ultimately selects for reversal of selumetinib resistance (revertant HT6244-R(-), right). This reversal of MEKi
resistance is due to loss of BRAF V600E amplification in these revertant cells and a re-setting of BRAF and p-ERK1/2 back to parental HT29
levels. P: phosphate group
these cells underwent substantial ERK1/2-dependent cell death that was partially caspase-dependent, i.e.,
apoptotic [Figure 3] . This cell death was associated with cleavage of the BH3-only protein BID to the
[11]
truncated pro-apoptotic form tBID, which is a consequence of CASP8 activation following stimulation of the
extrinsic pathway of apoptosis. In addition hyperactivation of ERK1/2 following MEKi removal promoted
expression of the pro-apoptotic BH3-only protein NOXA. Both tBID and NOXA inhibit pro-survival BCL2
family proteins at the outer mitochondrial membrane to promote the intrinsic pathway of apoptosis. That
BID cleavage occurred prior to PARP cleavage, a known CASP3 target, suggests that ERK1/2 hyperactivation
triggered the extrinsic pathway of apoptosis and activation of CASP8 prior to activation of the intrinsic
pathway and CASP3 . Pro-apoptotic NOXA undoubtedly contributes to activation of the intrinsic apoptotic
[11]
cascade but can also promote autophagy in response to high ERK1/2 activity by binding to MCL1 and
causing release of Beclin-1 [29,30] . Indeed, MEKi withdrawal from HT6244-R cells increased processing of LC3,
consistent with an upregulation of autophagy [Figure 3] . Thus HT6244-R cells exhibited hallmarks of both
[11]
apoptotic and autophagic cell death following selumetinib withdrawal, which almost certainly impose the
selection pressure that ultimately drives reversal of resistance. Thus, although COLO205 and HT29 tumour
cells have disabled multiple tumour suppressive mechanisms, including p53, and adapted to aberrant ERK1/2