
Page 92 - Read Online
P. 92
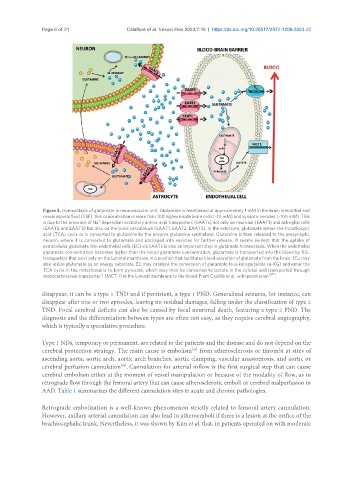
Page 6 of 21 Calafiore et al. Vessel Plus 2023;7:18 https://dx.doi.org/10.20517/2574-1209.2023.42
Figure 3. Homeostasis of glutamate in neurovascular unit. Glutamate is maintained at approximately 1 mM in the brain interstitial and
cerebrospinal fluid (CSF); this concentration is more than 100 higher inside brain cells (~10 mM) and synaptic vesicles (~100 mM). This
+
is due to the presence of Na -dependent excitatory amino-acid transporters (EAATs) not only on neuronal (EAAT1) and astroglial cells
(EAAT2 and EAAT3) but also on the brain vasculature (EAAT1, EAAT2, EAAT3). In the astrocyte, glutamate enters the tricarboxylic
acid (TCA) cycle or is converted to glutamine by the enzyme glutamine synthetase. Glutamine is then released to the presynaptic
neuron, where it is converted to glutamate and packaged into vesicles for further release. It seems evident that the uptake of
extracellular glutamate into endothelial cells (EC) via EAATs is also an important step in glutamate homeostasis. When the endothelial
glutamate concentration becomes higher than the blood glutamate concentration, glutamate is transported into the blood by XG-
transporters that exist only on the luminal membrane in a position that facilitates blood excretion of glutamate from the brain. ECs may
also utilize glutamate as an energy substrate. EC may catalyze the conversion of glutamate to α-ketoglutarate (α-KG) and enter the
TCA cycle in the mitochondria to form pyruvate, which may then be converted to lactate in the cytosol and transported through
[129]
monocarboxylate transporter 1 (MCT-1) in the luminal membrane to the blood. From Castillo et al., with permission .
disappear, it can be a type 1 TND and if persistent, a type 1 PND. Generalized seizures, for instance, can
disappear after one or two episodes, leaving no residual damages, falling under the classification of type 2
TND. Focal cerebral deficits can also be caused by focal neuronal death, featuring a type 2 PND. The
diagnosis and the differentiation between types are often not easy, as they require cerebral angiography,
which is typically a speculative procedure.
Type 1 NDs, temporary or permanent, are related to the patients and the disease and do not depend on the
[33]
cerebral protection strategy. The main cause is embolism from atherosclerosis or thrombi at sites of
ascending aorta, aortic arch, aortic arch branches, aortic clamping, vascular anastomosis, and aortic or
[34]
cerebral perfusion cannulation . Cannulation for arterial inflow is the first surgical step that can cause
cerebral embolism either at the moment of vessel manipulation or because of the modality of flow, as in
retrograde flow through the femoral artery that can cause atherosclerotic emboli or cerebral malperfusion in
AAD. Table 1 summarizes the different cannulation sites in acute and chronic pathologies.
Retrograde embolization is a well-known phenomenon strictly related to femoral artery cannulation.
However, axillary arterial cannulation can also lead to atheroemboli if there is a lesion at the orifice of the
brachiocephalic trunk. Nevertheless, it was shown by Kim et al. that, in patients operated on with moderate