
Page 10 - Read Online
P. 10
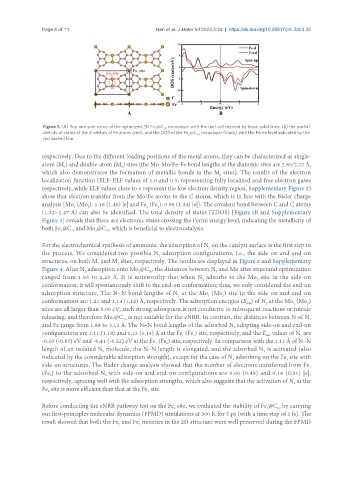
Page 4 of 11 Han et al. J Mater Inf 2023;3:24 https://dx.doi.org/10.20517/jmi.2023.32
Figure 1. (A) Top and side views of the optimized 2D Fe @C monolayer with the unit cell marked by black solid lines; (B) the partial
3
20
density of states of the d orbitals of Fe atoms (red), and the DOS of the Fe @C monolayer (black) with the Fermi level indicated by the
20
3
red dashed line.
respectively. Due to the different loading positions of the metal atoms, they can be characterized as single-
atom (M ) and double-atom (M ) sites (the Mo-Mo/Fe-Fe bond lengths at the diatomic sites are 2.85/2.22 Å,
1
2
which also demonstrates the formation of metallic bonds in the M sites). The results of the electron
2
localization function (ELF: ELF values of 1.0 and 0.5, representing fully localized and free electron gases
respectively, while ELF values close to 0 represent the low electron density region, Supplementary Figure 2)
show that electron transfer from the Mo/Fe atoms to the C atoms, which is in line with the Bader charge
analysis [Mo (Mo ): 1.16 (1.46) |e| and Fe (Fe ): 0.96 (1.34) |e|]. The covalent bond between C and C atoms
1
1
2
2
(1.32~1.47 Å) can also be identified. The total density of states (TDOS) [Figure 1B and Supplementary
Figure 3] reveals that there are electronic states crossing the Fermi energy level, indicating the metallicity of
both Fe @C and Mo @C , which is beneficial to electrocatalysis.
3
20
20
3
For the electrochemical synthesis of ammonia, the adsorption of N on the catalyst surface is the first step in
2
the process. We considered two possible N adsorption configurations, i.e., the side-on and end-on
2
structures, on both M and M sites, respectively. The results are displayed in Figure 2 and Supplementary
1
2
Figure 4. After N adsorption onto Mo @C , the distances between N and Mo after structural optimization
2
20
3
2
ranged from 1.95 to 2.25 Å. It is noteworthy that when N adsorbs to the Mo site in the side-on
2
2
conformation, it will spontaneously shift to the end-on conformation; thus, we only considered the end-on
adsorption structure. The N−N bond lengths of N at the Mo (Mo ) site in the side-on and end-on
2
1
2
conformations are 1.21 and 1.14 (1.12) Å, respectively. The adsorption energies (E ) of N at the Mo (Mo )
ads
2
2
1
sites are all larger than 5.00 eV; such strong adsorption is not conducive to subsequent reactions or nitride
releasing, and therefore Mo @C is not suitable for the eNRR. In contrast, the distances between N of N
3
2
20
and Fe range from 1.88 to 3.13 Å. The N−N bond lengths of the adsorbed N adopting side-on and end-on
2
configurations are 1.11 (1.18) and 1.13 (1.14) Å at the Fe (Fe ) site, respectively, and the E values of N are
2
1
2
ads
-0.10 (-0.57) eV and -0.41 (-0.52) eV at the Fe (Fe ) site, respectively. In comparison with the 1.11 Å of N−N
1
2
length of an isolated N molecule, the N−N length is elongated, and the adsorbed N is activated (also
2
2
indicated by the considerable adsorption strength), except for the case of N adsorbing on the Fe site with
2
1
side-on structures. The Bader charge analysis showed that the number of electrons transferred from Fe
1
(Fe ) to the adsorbed N with side-on and end-on configurations are 0.00 (0.49) and 0.16 (0.31) |e|,
2
2
respectively, agreeing well with the adsorption strengths, which also suggests that the activation of N at the
2
Fe site is more efficient than that at the Fe site.
1
2
Before conducting the eNRR pathway test on the Fe site, we evaluated the stability of Fe @C by carrying
2
3
20
out first-principles molecular dynamics (FPMD) simulations at 300 K for 5 ps (with a time step of 1 fs). The
result showed that both the Fe and Fe moieties in the 2D structure were well preserved during the FPMD
2
1