
Page 64 - Read Online
P. 64
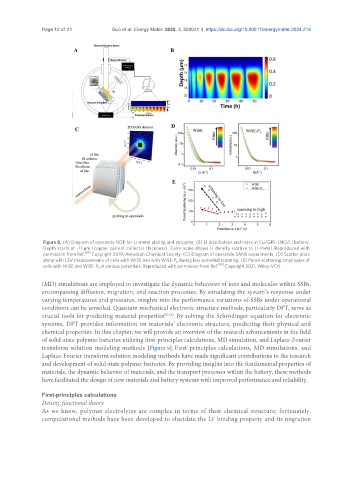
Page 12 of 21 Guo et al. Energy Mater. 2025, 5, 500041 https://dx.doi.org/10.20517/energymater.2024.214
Figure 8. (A) Diagram of operando NDP for Li-metal plating and stripping. (B) Li distribution and mass in Cu/GPE-LNO/Li battery.
Depth starts at ~11 μm (copper current collector thickness). Color scale shows Li density relative to Li-metal. Reproduced with
[83]
permission from Ref. Copyright 2019, American Chemical Society. (C) Diagram of operando SANS experiments. (D) Scatter plots
along with LSV measurements of cells with WiSE and with WiSE-P during low-potential scanning. (E) Porod scattering amplitudes of
5
cells with WiSE and WiSE-P at various potentials. Reproduced with permission from Ref. [90] Copyright 2021, Wiley-VCH.
5
(MD) simulations are employed to investigate the dynamic behaviors of ions and molecules within SSBs,
encompassing diffusion, migration, and reaction processes. By simulating the system’s response under
varying temperatures and pressures, insights into the performance variations of SSBs under operational
conditions can be unveiled. Quantum mechanical electronic structure methods, particularly DFT, serve as
crucial tools for predicting material properties [91,92] . By solving the Schrödinger equation for electronic
systems, DFT provides information on materials’ electronic structure, predicting their physical and
chemical properties. In this chapter, we will provide an overview of the research advancements in the field
of solid-state polymer batteries utilizing first-principles calculations, MD simulation, and Laplace-Fourier
transform solution modeling methods [Figure 9]. First-principles calculations, MD simulations, and
Laplace-Fourier transform solution modeling methods have made significant contributions to the research
and development of solid-state polymer batteries. By providing insights into the fundamental properties of
materials, the dynamic behavior of materials, and the transport processes within the battery, these methods
have facilitated the design of new materials and battery systems with improved performance and reliability.
First-principles calculations
Density functional theory
As we know, polymer electrolytes are complex in terms of their chemical structure; fortunately,
+
computational methods have been developed to elucidate the Li binding property and its migration