
Page 134 - Read Online
P. 134
Xu et al. Chem Synth 2023;3:17 https://dx.doi.org/10.20517/cs.2022.35 Page 7 of 10
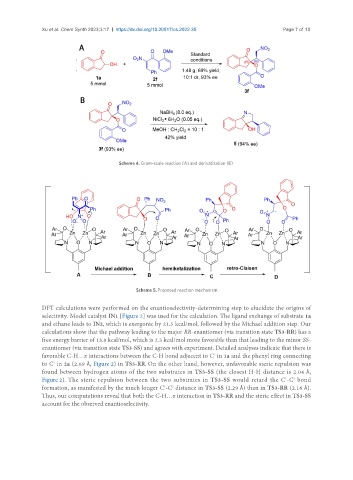
Scheme 4. Gram-scale reaction (A) and derivatization (B).
Scheme 5. Proposed reaction mechanism.
DFT calculations were performed on the enantioselectivity-determining step to elucidate the origins of
selectivity. Model catalyst IN1 [Figure 2] was used for the calculation. The ligand exchange of substrate 1a
and ethane leads to IN2, which is exergonic by 21.5 kcal/mol, followed by the Michael addition step. Our
calculations show that the pathway leading to the major RR-enantiomer (via transition state TS3-RR) has a
free energy barrier of 13.8 kcal/mol, which is 2.3 kcal/mol more favorable than that leading to the minor SS-
enantiomer (via transition state TS3-SS) and agrees with experiment. Detailed analyses indicate that there is
favorable C-H…π interactions between the C-H bond adjacent to C in 1a and the phenyl ring connecting
1
to C in 2a (2.69 Å, Figure 2) in TS3-RR. On the other hand, however, unfavorable steric repulsion was
2
found between hydrogen atoms of the two substrates in TS3-SS (the closest H-H distance is 2.04 Å,
1
2
Figure 2). The steric repulsion between the two substrates in TS3-SS would retard the C -C bond
formation, as manifested by the much longer C -C distance in TS3-SS (2.29 Å) than in TS3-RR (2.16 Å).
2
1
Thus, our computations reveal that both the C-H…π interaction in TS3-RR and the steric effect in TS3-SS
account for the observed enantioselectivity.