
Page 20 - Read Online
P. 20
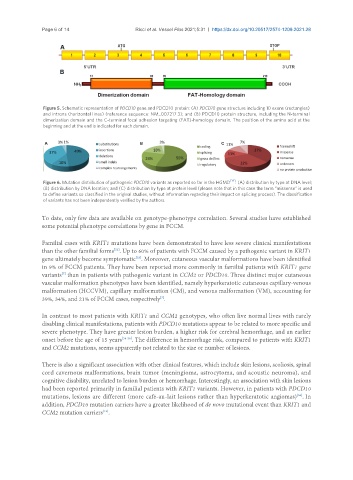
Page 6 of 14 Ricci et al. Vessel Plus 2021;5:31 https://dx.doi.org/10.20517/2574-1209.2021.28
Figure 5. Schematic representation of PDCD10 gene and PDCD10 protein: (A) PDCD10 gene structure including 10 exons (rectangles)
and introns (horizontal lines) (reference sequence: NM_007217.3); and (B) PDCD10 protein structure, including the N-terminal
dimerization domain and the C-terminal focal adhesion targeting (FAT)-homology domain. The position of the amino acid at the
beginning and at the end is indicated for each domain.
Figure 6. Mutation distribution of pathogenic PDCD10 variants as reported so far in the HGMD [14] : (A) distribution by type at DNA level;
(B) distribution by DNA location; and (C) distribution by type at protein level (please note that in this case the term “missense” is used
to define variants so classified in the original studies, without information regarding their impact on splicing process). The classification
of variants has not been independently verified by the authors.
To date, only few data are available on genotype-phenotype correlation. Several studies have established
some potential phenotype correlations by gene in FCCM.
Familial cases with KRIT1 mutations have been demonstrated to have less severe clinical manifestations
[52]
than the other familial forms . Up to 60% of patients with FCCM caused by a pathogenic variant in KRIT1
gene ultimately become symptomatic . Moreover, cutaneous vascular malformations have been identified
[53]
in 9% of FCCM patients. They have been reported more commonly in familial patients with KRIT1 gene
variants than in patients with pathogenic variant in CCM2 or PDCD10. Three distinct major cutaneous
[7]
vascular malformation phenotypes have been identified, namely hyperkeratotic cutaneous capillary-venous
malformation (HCCVM), capillary malformation (CM), and venous malformation (VM), accounting for
39%, 34%, and 21% of FCCM cases, respectively .
[7]
In contrast to most patients with KRIT1 and CCM2 genotypes, who often live normal lives with rarely
disabling clinical manifestations, patients with PDCD10 mutations appear to be related to more specific and
severe phenotype. They have greater lesion burden, a higher risk for cerebral hemorrhage, and an earlier
onset before the age of 15 years [54-56] . The difference in hemorrhage risk, compared to patients with KRIT1
and CCM2 mutations, seems apparently not related to the size or number of lesions.
There is also a significant association with other clinical features, which include skin lesions, scoliosis, spinal
cord cavernous malformations, brain tumor (meningioma, astrocytoma, and acoustic neuroma), and
cognitive disability, unrelated to lesion burden or hemorrhage. Interestingly, an association with skin lesions
had been reported primarily in familial patients with KRIT1 variants. However, in patients with PDCD10
[56]
mutations, lesions are different (more café-au-lait lesions rather than hyperkeratotic angiomas) . In
addition, PDCD10 mutation carriers have a greater likelihood of de novo mutational event than KRIT1 and
CCM2 mutation carriers .
[13]