
Page 11 - Read Online
P. 11
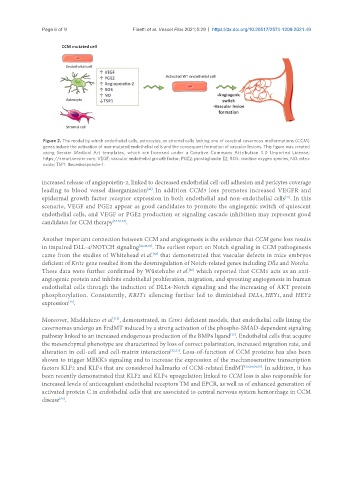
Page 6 of 9 Finetti et al. Vessel Plus 2021;5:29 https://dx.doi.org/10.20517/2574-1209.2021.49
Figure 2. The model by which endothelial cells, astrocytes, or stromal cells lacking one of cerebral cavernous malformations (CCM)
genes induce the activation of non-mutated endothelial cells and the consequent formation of vascular lesions. This figure was created
using Servier Medical Art templates, which are licensed under a Creative Commons Attribution 3.0 Unported License;
https://smart.servier.com. VEGF: vascular endothelial growth factor; PGE2: prostaglandin E2; ROS: reactive oxygen species; NO: nitric
oxide; TSP1: thrombospondin-1.
increased release of angiopoietin-2, linked to decreased endothelial cell-cell adhesion and pericytes coverage
[46]
leading to blood vessel disorganization . In addition CCM3 loss promotes increased VEGFR and
epidermal growth factor receptor expression in both endothelial and non-endothelial cells . In this
[47]
scenario, VEGF and PGE2 appear as good candidates to promote the angiogenic switch of quiescent
endothelial cells, and VEGF or PGE2 production or signaling cascade inhibition may represent good
candidates for CCM therapy [37,38,48] .
Another important connection between CCM and angiogenesis is the evidence that CCM gene loss results
in impaired DLL-4/NOTCH signaling [40,49,50] . The earliest report on Notch signaling in CCM pathogenesis
came from the studies of Whitehead et al. that demonstrated that vascular defects in mice embryos
[52]
deficient of Krit1 gene resulted from the downregulation of Notch-related genes including Dll4 and Notch4.
These data were further confirmed by Wüstehube et al. which reported that CCM1 acts as an anti-
[40]
angiogenic protein and inhibits endothelial proliferation, migration, and sprouting angiogenesis in human
endothelial cells through the induction of DLL4-Notch signaling and the increasing of AKT protein
phosphorylation. Consistently, KRIT1 silencing further led to diminished DLL4, HEY1, and HEY2
[40]
expression .
Moreover, Maddaluno et al. , demonstrated, in Ccm1 deficient models, that endothelial cells lining the
[15]
cavernomas undergo an EndMT induced by a strong activation of the phospho-SMAD-dependent signaling
[15]
pathway linked to an increased endogenous production of the BMP6 ligand . Endothelial cells that acquire
the mesenchymal phenotype are characterized by loss of correct polarization, increased migration rate, and
alteration in cell-cell and cell-matrix interactions [52,53] . Loss-of-function of CCM proteins has also been
shown to trigger MEKK3 signaling and to increase the expression of the mechanosensitive transcription
factors KLF2 and KLF4 that are considered hallmarks of CCM-related EndMT [18,24,54,55] . In addition, it has
been recently demonstrated that KLF2 and KLF4 upregulation linked to CCM loss is also responsible for
increased levels of anticoagulant endothelial receptors TM and EPCR, as well as of enhanced generation of
activated protein C in endothelial cells that are associated to central nervous system hemorrhage in CCM
disease .
[56]