
Page 10 - Read Online
P. 10
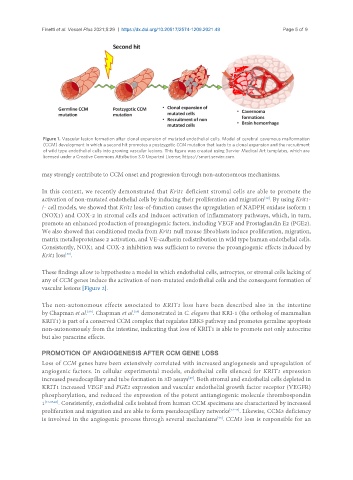
Finetti et al. Vessel Plus 2021;5:29 https://dx.doi.org/10.20517/2574-1209.2021.49 Page 5 of 9
Figure 1. Vascular lesion formation after clonal expansion of mutated endothelial cells. Model of cerebral cavernous malformation
(CCM) development in which a second hit promotes a postzygotic CCM mutation that leads to a clonal expansion and the recruitment
of wild type endothelial cells into growing vascular lesions. This figure was created using Servier Medical Art templates, which are
licensed under a Creative Commons Attribution 3.0 Unported License; https://smart.servier.com.
may strongly contribute to CCM onset and progression through non-autonomous mechanisms.
In this context, we recently demonstrated that Krit1 deficient stromal cells are able to promote the
[38]
activation of non-mutated endothelial cells by inducing their proliferation and migration . By using Krit1-
/- cell models, we showed that Krit1 loss-of-function causes the upregulation of NADPH oxidase isoform 1
(NOX1) and COX-2 in stromal cells and induces activation of inflammatory pathways, which, in turn,
promote an enhanced production of proangiogenic factors, including VEGF and Prostaglandin E2 (PGE2).
We also showed that conditioned media from Krit1 null mouse fibroblasts induce proliferation, migration,
matrix metalloproteinase 2 activation, and VE-cadherin redistribution in wild type human endothelial cells.
Consistently, NOX1 and COX-2 inhibition was sufficient to reverse the proangiogenic effects induced by
Krit1 loss .
[38]
These findings allow to hypothesize a model in which endothelial cells, astrocytes, or stromal cells lacking of
any of CCM genes induce the activation of non-mutated endothelial cells and the consequent formation of
vascular lesions [Figure 2].
The non-autonomous effects associated to KRIT1 loss have been described also in the intestine
by Chapman et al. . Chapman et al. demonstrated in C. elegans that KRI-1 (the ortholog of mammalian
[39]
[39]
KRIT1) is part of a conserved CCM complex that regulates ERK5 pathway and promotes germline apoptosis
non-autonomously from the intestine, indicating that loss of KRIT1 is able to promote not only autocrine
but also paracrine effects.
PROMOTION OF ANGIOGENESIS AFTER CCM GENE LOSS
Loss of CCM genes have been extensively correlated with increased angiogenesis and upregulation of
angiogenic factors. In cellular experimental models, endothelial cells silenced for KRIT1 expression
[40]
increased pseudocapillary and tube formation in 3D assays . Both stromal and endothelial cells depleted in
KRIT1 increased VEGF and PGE2 expression and vascular endothelial growth factor receptor (VEGFR)
phosphorylation, and reduced the expression of the potent antiangiogenic molecule thrombospondin
1 [17,38,41] . Consistently, endothelial cells isolated from human CCM specimens are characterized by increased
proliferation and migration and are able to form pseudocapillary networks [42-44] . Likewise, CCM3 deficiency
[45]
is involved in the angiogenic process through several mechanisms . CCM3 loss is responsible for an