
Page 45 - Read Online
P. 45
Page 117 Waller et al. J Transl Genet Genom 2021;5:112-23 I http://dx.doi.org/10.20517/jtgg.2021.09
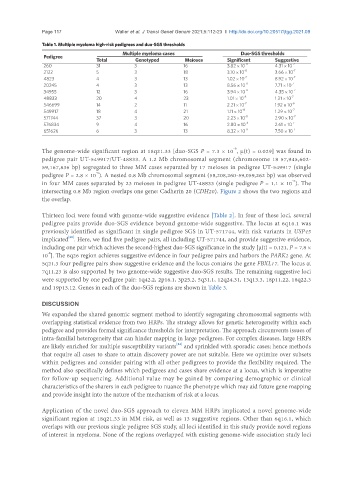
Table 1. Multiple myeloma high-risk pedigrees and duo-SGS thresholds
Multiple myeloma cases Duo-SGS thresholds
Pedigree
Total Genotyped Meioses Significant Suggestive
260 31 3 16 3.82 × 10 -8 4.31 × 10 -7
2122 5 3 18 3.10 × 10 -8 3.66 × 10 -7
4823 4 3 13 1.02 × 10 -7 8.92 × 10 -7
20245 4 3 13 8.56 × 10 -8 7.71 × 10 -7
34955 12 3 16 3.94 × 10 -8 4.35 × 10 -7
48833 20 4 23 1.01 × 10 -8 1.21 × 10 -7
546699 14 2 11 2.21 × 10 -7 1.92 × 10 -6
549917 18 4 21 1.11 × 10 -8 1.29 × 10 -7
571744 37 3 20 2.23 × 10 -8 2.90 × 10 -7
576834 9 4 16 2.80 × 10 -8 2.61 × 10 -7
651626 6 3 13 8.32 × 10 -8 7.50 × 10 -7
-9
The genome-wide significant region at 18q21.33 [duo-SGS P = 7.3 × 10 , µ(t) = 0.029] was found in
pedigree pair UT-549917/UT-48833. A 1.2 Mb chromosomal segment (chromosome 18 57,945,602-
59,167,836 bp) segregated to three MM cases separated by 17 meioses in pedigree UT-549917 (single
-5
pedigree P = 2.8 × 10 ). A nested 0.8 Mb chromosomal segment (58,208,260-59,059,262 bp) was observed
-5
in four MM cases separated by 23 meioses in pedigree UT-48833 (single pedigree P = 1.1 × 10 ). The
intersecting 0.8 Mb region overlaps one gene: Cadherin 20 (CDH20). Figure 2 shows the two regions and
the overlap.
Thirteen loci were found with genome-wide suggestive evidence [Table 2]. In four of these loci, several
pedigree pairs provide duo-SGS evidence beyond genome-wide suggestive. The locus at 6q16.1 was
previously identified as significant in single pedigree SGS in UT-571744, with risk variants in USP45
[20]
implicated . Here, we find five pedigree pairs, all including UT-571744, and provide suggestive evidence,
including one pair which achieves the second-highest duo-SGS significance in the study [µ(t) = 0.121, P = 7.8 ×
-8
10 ]. The 6q26 region achieves suggestive evidence in four pedigree pairs and harbors the PARK2 gene. At
5q21.3 four pedigree pairs show suggestive evidence and the locus contains the gene FBXL17. The locus at
7q11.23 is also supported by two genome-wide suggestive duo-SGS results. The remaining suggestive loci
were supported by one pedigree pair: 1q42.2, 2p16.1, 3p25.2, 5q31.1, 12q24.31, 13q13.3, 18p11.22, 18q22.3
and 19p13.12. Genes in each of the duo-SGS regions are shown in Table 3.
DISCUSSION
We expanded the shared genomic segment method to identify segregating chromosomal segments with
overlapping statistical evidence from two HRPs. The strategy allows for genetic heterogeneity within each
pedigree and provides formal significance thresholds for interpretation. The approach circumvents issues of
intra-familial heterogeneity that can hinder mapping in large pedigrees. For complex diseases, large HRPs
are likely enriched for multiple susceptibility variants and sprinkled with sporadic cases; hence methods
[24]
that require all cases to share to attain discovery power are not suitable. Here we optimize over subsets
within pedigrees and consider pairing with all other pedigrees to provide the flexibility required. The
method also specifically defines which pedigrees and cases share evidence at a locus, which is imperative
for follow-up sequencing. Additional value may be gained by comparing demographic or clinical
characteristics of the sharers in each pedigree to nuance the phenotype which may aid future gene mapping
and provide insight into the nature of the mechanism of risk at a locus.
Application of the novel duo-SGS approach to eleven MM HRPs implicated a novel genome-wide
significant region at 18q21.33 in MM risk, as well as 13 suggestive regions. Other than 6q16.1, which
overlaps with our previous single pedigree SGS study, all loci identified in this study provide novel regions
of interest in myeloma. None of the regions overlapped with existing genome-wide association study loci