
Page 585 - Read Online
P. 585
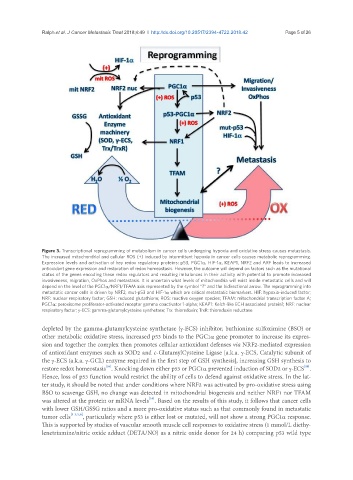
Ralph et al. J Cancer Metastasis Treat 2018;4:49 I http://dx.doi.org/10.20517/2394-4722.2018.42 Page 5 of 26
Figure 3. Transcriptional reprogramming of metabolism in cancer cells undergoing hypoxia and oxidative stress causes metastasis.
The increased mitochondrial and cellular ROS (+) induced by intermittent hypoxia in cancer cells causes metabolic reprogramming.
Expression levels and activation of key redox regulatory proteins: p53, PGC1α, HIF-1α, KEAP1, NRF2 and ARF leads to increased
antioxidant gene expression and restoration of redox homeostasis. However, the outcome will depend on factors such as the mutational
status of the genes encoding these redox regulators and resulting imbalances in their activity with potential to promote increased
invasiveness, migration, OxPhos and metastasis. It is uncertain what levels of mitochondria will exist inside metastatic cells and will
depend on the level of the PGC1α/NRF1/TFAM axis represented by the symbol “?” and the bidirectional arrow. The reprogramming into
metastatic cancer cells is driven by NRF2, mut-p53 and HIF-1α which are critical metastatic biomarkers. HIF: hypoxia-induced factor;
NRF: nuclear respiratory factor; GSH: reduced glutathione; ROS: reactive oxygen species; TFAM: mitochondrial transcription factor A;
PGC1α: peroxisome proliferator-activated receptor gamma coactivator 1-alpha; KEAP1: Kelch-like ECH associated protein1; NRF: nuclear
respiratory factor; γ-ECS: gamma-glutamylcysteine synthetase; Trx: thioredoxin; TrxR: thioredoxin reductase
depleted by the gamma-glutamylcysteine synthetase (γ-ECS) inhibitor, buthionine sulfoximine (BSO) or
other metabolic oxidative stress, increased p53 binds to the PGC1α gene promoter to increase its expres-
sion and together the complex then promotes cellular antioxidant defenses via NRF2-mediated expression
of antioxidant enzymes such as SOD2 and c-GlutamylCysteine Ligase [a.k.a. γ-ECS, Catalytic subunit of
the γ-ECS (a.k.a. γ-GCL) enzyme required in the first step of GSH synthesis], increasing GSH synthesis to
[16]
[16]
restore redox homeostasis . Knocking down either p53 or PGC1α prevented induction of SOD2 or γ-ECS .
Hence, loss of p53 function would restrict the ability of cells to defend against oxidative stress. In the lat-
ter study, it should be noted that under conditions where NRF2 was activated by pro-oxidative stress using
BSO to scavenge GSH, no change was detected in mitochondrial biogenesis and neither NRF1 nor TFAM
[16]
was altered at the protein or mRNA levels . Based on the results of this study, it follows that cancer cells
with lower GSH/GSSG ratios and a more pro-oxidative status such as that commonly found in metastatic
tumor cells [1-3,5,6] , particularly where p53 is either lost or mutated, will not show a strong PGC1α response.
This is supported by studies of vascular smooth muscle cell responses to oxidative stress (1 mmol/L diethy-
lenetriamine/nitric oxide adduct (DETA/NO) as a nitric oxide donor for 24 h) comparing p53 wild type