
Page 12 - Read Online
P. 12
Page 6 of 14 Gottlieb et al. J Cancer Metastasis Treat 2018;4:37 I http://dx.doi.org/10.20517/2394-4722.2018.26
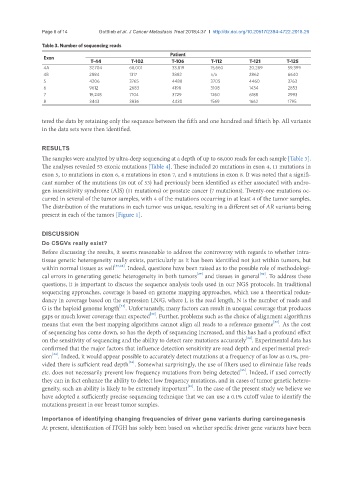
Table 3. Number of sequencing reads
Patient
Exon
T-44 T-102 T-106 T-112 T-121 T-125
4A 37,704 68,001 33,819 15,660 20,289 59,399
4B 2884 1317 3882 n/a 2862 6640
5 4206 3765 4488 3705 4460 3763
6 9612 2683 4198 3108 1434 2853
7 19,248 7104 3729 1260 6188 2993
8 3443 3836 4430 1569 1662 1795
tered the data by retaining only the sequence between the fifth and one hundred and fiftieth bp. All variants
in the data sets were then identified.
RESULTS
The samples were analyzed by ultra-deep sequencing at a depth of up to 68,000 reads for each sample [Table 3].
The analyses revealed 53 exonic mutations [Table 4]. These included 20 mutations in exon 4, 11 mutations in
exon 5, 10 mutations in exon 6, 4 mutations in exon 7, and 8 mutations in exon 8. It was noted that a signifi-
cant number of the mutations (18 out of 53) had previously been identified as either associated with andro-
gen insensitivity syndrome (AIS) (11 mutations) or prostate cancer (7 mutations). Twenty-one mutations oc-
curred in several of the tumor samples, with 4 of the mutations occurring in at least 4 of the tumor samples.
The distribution of the mutations in each tumor was unique, resulting in a different set of AR variants being
present in each of the tumors [Figure 1].
DISCUSSION
Do CSGVs really exist?
Before discussing the results, it seems reasonable to address the controversy with regards to whether intra-
tissue genetic heterogeneity really exists, particularly as it has been identified not just within tumors, but
within normal tissues as well [27,28] . Indeed, questions have been raised as to the possible role of methodologi-
[29]
[30]
cal errors in generating genetic heterogeneity in both tumors and tissues in general . To address these
questions, it is important to discuss the sequence analysis tools used in our NGS protocols. In traditional
sequencing approaches, coverage is based on genome mapping approaches, which use a theoretical redun-
dancy in coverage based on the expression LN/G, where L is the read length, N is the number of reads and
[31]
G is the haploid genome length . Unfortunately, many factors can result in unequal coverage that produces
[32]
gaps or much lower coverage than expected . Further, problems such as the choice of alignment algorithms
[33]
means that even the best mapping algorithms cannot align all reads to a reference genome . As the cost
of sequencing has come down, so has the depth of sequencing increased, and this has had a profound effect
[34]
on the sensitivity of sequencing and the ability to detect rare mutations accurately . Experimental data has
confirmed that the major factors that influence detection sensitivity are read depth and experimental preci-
[34]
sion . Indeed, it would appear possible to accurately detect mutations at a frequency of as low as 0.1%, pro-
[34]
vided there is sufficient read depth . Somewhat surprisingly, the use of filters used to eliminate false reads
[35]
etc. does not necessarily prevent low frequency mutations from being detected . Indeed, if used correctly
they can in fact enhance the ability to detect low frequency mutations, and in cases of tumor genetic hetero-
[35]
geneity, such an ability is likely to be extremely important . In the case of the present study we believe we
have adopted a sufficiently precise sequencing technique that we can use a 0.1% cutoff value to identify the
mutations present in our breast tumor samples.
Importance of identifying changing frequencies of driver gene variants during carcinogenesis
At present, identification of ITGH has solely been based on whether specific driver gene variants have been