
Page 10 - Read Online
P. 10
Page 4 of 14 Gottlieb et al. J Cancer Metastasis Treat 2018;4:37 I http://dx.doi.org/10.20517/2394-4722.2018.26
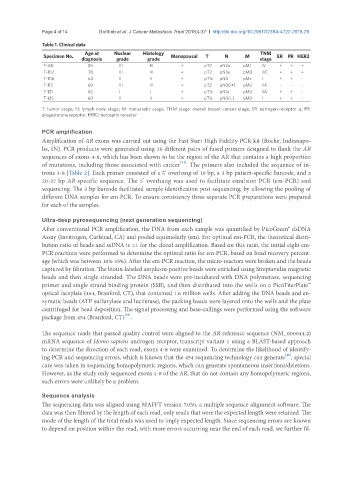
Table 1. Clinical data
Specimen No. Age at Nuclear Histology Menopausal T N M TNM ER PR HER2
diagnosis grade grade stage
T-44 55 III III + pT2 pN2a pM1 IV + + +
T-102 78 III III + pT2 pN3a pM0 IIIC + + +
T-106 64 II II + pT1c pN0 pMx I + + -
T-112 60 III III + pT2 pN0(i+) pM0 IIA - - -
T-121 62 I I + pT1c pN1a pM0 IIA + + -
T-125 60 II II + pT1c pN0(i-) pM0 I + + -
T: tumor stage; N: lymph node stage; M: metastatic stage; TNM stage: overall breast cancer stage; ER: estrogen receptor a; PR:
progesterone receptor; HER2: herceptin receptor
PCR amplification
Amplification of AR exons was carried out using the Fast Start High Fidelity PCR kit (Roche, Indianapo-
lis, IN). PCR products were generated using 36 different pairs of fused primers designed to flank the AR
sequences of exons 4-8, which has been shown to be the region of the AR that contains a high proportion
[24]
of mutations, including those associated with cancer . The primers also included the sequence of in-
trons 3-8 [Table 2]. Each primer consisted of a 5’ overhang of 19 bp, a 3 bp patient-specific barcode, and a
20-27 bp AR-specific sequence. The 5’ overhang was used to facilitate emulsion PCR (em-PCR) and
sequencing. The 3 bp barcode facilitated sample identification post sequencing, by allowing the pooling of
different DNA samples for em-PCR. To ensure consistency three separate PCR preparations were prepared
for each of the samples.
Ultra-deep pyrosequencing (next generation sequencing)
After conventional PCR amplification, the DNA from each sample was quantified by PicoGreen® dsDNA
Assay (Invitrogen, Carlsbad, CA) and pooled equimolarly (em). For optimal em-PCR, the theoretical distri-
bution ratio of beads and ssDNA is 1:1 for the clonal amplification. Based on this ratio, the initial eight em-
PCR reactions were performed to determine the optimal ratio for em-PCR, based on bead recovery percent-
age (which was between 10%-15%). After the em-PCR reaction, the micro-reactors were broken and the beads
captured by filtration. The biotin-labeled amplicon-positive beads were enriched using Streptavidin magnetic
beads and then single stranded. The DNA beads were pre-incubated with DNA polymerase, sequencing
primer and single strand binding protein (SSB), and then distributed into the wells on a PicoTiterPlate™
optical faceplate (454, Branford, CT), that contained 1.6 million wells. After adding the DNA beads and en-
zymatic beads (ATP sulfurylase and luciferase), the packing beads were layered onto the wells and the plate
centrifuged for bead deposition. The signal processing and base-callings were performed using the software
[25]
package from 454 (Branford, CT) .
The sequence reads that passed quality control were aligned to the AR reference sequence (NM_000044.2)
mRNA sequence of Homo sapiens androgen receptor, transcript variant 1 using a BLAST-based approach
to determine the direction of each read; exons 4-8 were examined. To determine the likelihood of identify-
[26]
ing PCR and sequencing errors, which is known that the 454 sequencing technology can generate , special
care was taken in sequencing homopolymeric regions, which can generate spontaneous insertions/deletions.
However, as the study only sequenced exons 4-8 of the AR, that do not contain any homopolymeric regions,
such errors were unlikely be a problem.
Sequence analysis
The sequencing data was aligned using MAFFT version 7.050, a multiple sequence alignment software. The
data was then filtered by the length of each read, only reads that were the expected length were retained. The
mode of the length of the total reads was used to imply expected length. Since sequencing errors are known
to depend on position within the read, with more errors occurring near the end of each read, we further fil-