
Page 937 - Read Online
P. 937
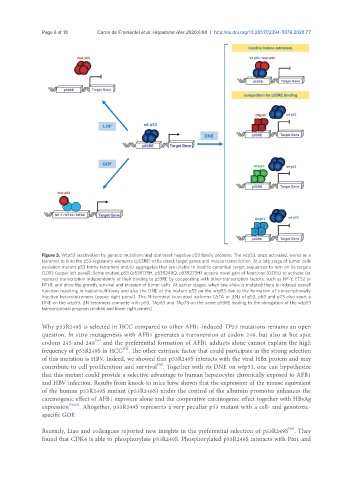
Page 6 of 18 Caron de Fromentel et al. Hepatoma Res 2020;6:80 I http://dx.doi.org/10.20517/2394-5079.2020.77
Figure 3. Wtp53 inactivation by genetic mutations and dominant negative p53 family proteins. The wtp53, once activated, works as a
tetramer to bind the p53-regulatory elements (p53RE) of its direct target genes and induce transcription. At a late stage of tumor cells
evolution mutant p53 forms tetramers and/or aggregates that are unable to bind to canonical target sequences to turn on its targets
(LOF) (upper left panel). Some mutant p53 (p53R175H, p53R248Q, p53R273H) acquire novel gain of functions (GOFs) to activate (or
repress) transcription independently of their binding to p53RE by cooperating with other transcription factors, such as NF-Y, ETS2 or
NFkB, and drive the growth, survival and invasion of tumor cells. At earlier stages, when one allele is mutated there is reduced overall
function resulting in haploinsufficieny and also the DNE of the mutant p53 on the wtp53 due to the formation of transcriptionally
inactive heterotetramers (upper right panel). The N-terminal truncated isoforms (ΔTA or ΔN) of p53, p63 and p73 also exert a
DNE on the wtp53. ΔN-tetramers compete with p53, TAp63 and TAp73 on the same p53RE leading to the abrogation of the wtp53
transcriptional program (middle and lower right panels)
Why p53R249S is selected in HCC compared to other AFB1-induced TP53 mutations remains an open
question. In vitro mutagenesis with AFB1 generates a transversion at codon 249, but also at hot spot
[44]
codons 245 and 248 and the preferential formation of AFB1 adducts alone cannot explain the high
[61]
frequency of p53R249S in HCC . The other extrinsic factor that could participate in the strong selection
of this mutation is HBV. Indeed, we showed that p53R249S interacts with the viral HBx protein and may
[58]
contribute to cell proliferation and survival . Together with its DNE on wtp53, one can hypothesize
that this mutant could provide a selective advantage to human hepatocytes chronically exposed to AFB1
and HBV infection. Results from knock-in mice have shown that the expression of the mouse equivalent
of the human p53R249S mutant (p53R246S) under the control of the albumin promoter enhances the
carcinogenic effect of AFB1 exposure alone and the cooperative carcinogenic effect together with HBsAg
expression [59,62] . Altogether, p53R249S represents a very peculiar p53 mutant with a cell- and genotoxic-
specific GOF.
[60]
Recently, Liao and colleagues reported new insights in the preferential selection of p53R249S . They
found that CDK4 is able to phosphorylate p53R249S. Phosphorylated p53R249S interacts with Pin1 and