
Page 82 - Read Online
P. 82
Page 168 Sulaiman et al. J Transl Genet Genom 2020;4:159-87 I https://doi.org/10.20517/jtgg.2020.27
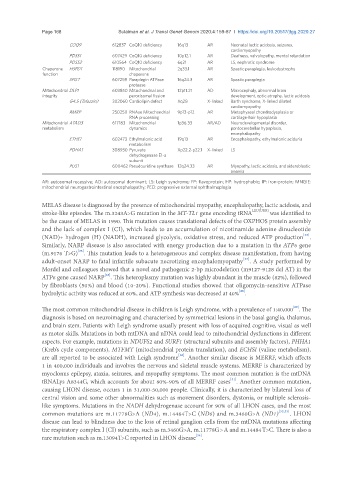
COQ9 612837 CoQ10 deficiency 16q13 AR Neonatal lactic acidosis, seizures,
cardiomyopathy
PDSS1 607429 CoQ10 deficiency 10p12.1 AR Deafness, valvulopathy, mental retardation
PDSS2 610564 CoQ10 deficiency 6q21 AR LS, nephrotic syndrome
Chaperone HSPD1 118190 Mitochondrial 2q33.1 AR Spastic paraplegia, leukodystrophy
function chaperone
SPG7 607259 Paraplegin ATPase 16q24.3 AR Spastic paraplegia
protease
Mitochondrial DLP1 603850 Mitochondrial and 12p11.21 AD Microcephaly, abnormal brain
integrity peroxisomal fission development, optic atrophy, lactic acidosis
G4.5 (Tafazzin) 302060 Cardiolipin defect Xq28 X-linked Barth syndrome, X-linked dilated
cardiomyopathy
RMRP 250250 RNAse Mitochondrial 9p13-p12 AR Metaphyseal chondrodysplasia or
RNA processing cartilage-hair hypoplasia
Mitochondrial ATAD3 617183 Mitochondrial 1p36.33 AR/AD Neurodevelopmental disorder,
metabolism dynamics pontocerebellar hypoplasia,
encephalopathy
ETHE1 602473 Ethylmalonic acid 19q13 AR Encephalopathy, ethylmalonic aciduria
metabolism
PDHA1 308930 Pyruvate Xp22.2-p22.1 X-linked LS
dehydrogenase E1-a
subunit
PUS1 600462 Pseudouridine synthase 12q24.33 AR Myopathy, lactic acidosis, and sideroblastic
anemia
AR: autosomal recessive; AD: autosomal dominant; LS: Leigh syndrome; FP: flavoprotein; HP: hydrophobic; IP: iron-protein; MNGIE:
mitochondrial neurogastrointestinal encephalopathy; PEO: progressive external ophthalmoplegia
MELAS disease is diagnosed by the presence of mitochondrial myopathy, encephalopathy, lactic acidosis, and
stroke-like episodes. The m.3243A>G mutation in the MT-TL1 gene encoding tRNA LEU(UUR) was identified to
be the cause of MELAS in 1990. This mutation causes translational defects of the OXPHOS protein assembly
and the lack of complex I (CI), which leads to an accumulation of nicotinamide adenine dinucleotide
(NAD)+ hydrogen (H) (NADH), increased glycolysis, oxidative stress, and reduced ATP production .
[25]
Similarly, NARP disease is also associated with energy production due to a mutation in the ATP6 gene
[26]
(m.9176 T>G) . This mutation leads to a heterogeneous and complex disease manifestation, from having
[27]
adult-onset NARP to fatal infantile subacute necrotizing encephalomyopathy . A study performed by
Mordel and colleagues showed that a novel and pathogenic 2-bp microdeletion (m9127-9128 del AT) in the
[28]
ATP6 gene caused NARP . This heteroplasmy mutation was highly abundant in the muscle (82%), followed
by fibroblasts (50%) and blood (10-20%). Functional studies showed that oligomycin-sensitive ATPase
[28]
hydrolytic activity was reduced at 60%, and ATP synthesis was decreased at 40% .
[29]
The most common mitochondrial disease in children is Leigh syndrome, with a prevalence of 1:40,000 . The
diagnosis is based on neuroimaging and characterized by symmetrical lesions in the basal ganglia, thalamus,
and brain stem. Patients with Leigh syndrome usually present with loss of acquired cognitive, visual as well
as motor skills. Mutations in both mtDNA and nDNA could lead to mitochondrial dysfunctions in different
aspects. For example, mutations in NDUFS2 and SURF1 (structural subunits and assembly factors), PHHA1
(Kreb’s cycle components), MTFMT (mitochondrial protein translation), and ECHSI (valine metabolism),
[30]
are all reported to be associated with Leigh syndrome . Another similar disease is MERRF, which affects
1 in 400,000 individuals and involves the nervous and skeletal muscle systems. MERRF is characterized by
myoclonus epilepsy, ataxia, seizures, and myopathy symptoms. The most common mutation is the mtDNA
[31]
tRNALys A8344G, which accounts for about 80%-90% of all MERRF cases . Another common mutation,
causing LHON disease, occurs 1 in 31,000-50,000 people. Clinically, it is characterized by bilateral loss of
central vision and some other abnormalities such as movement disorders, dystonia, or multiple sclerosis-
like symptoms. Mutations in the NADH dehydrogenase account for 90% of all LHON cases, and the most
common mutations are m.11778G>A (ND4), m.14484T>C (ND6) and m.3460G>A (ND1) [32,33] . LHON
disease can lead to blindness due to the loss of retinal ganglion cells from the mtDNA mutations affecting
the respiratory complex I (CI) subunits, such as m.3460G>A, m.11778G > A and m.14484 T>C. There is also a
[34]
rare mutation such as m.13094T>C reported in LHON disease .