
Page 30 - Read Online
P. 30
Page 345 Gonzalez Castillo et al. J Transl Genet Genom. 2025;9:338-51 https://dx.doi.org/10.20517/jtgg.2025.57
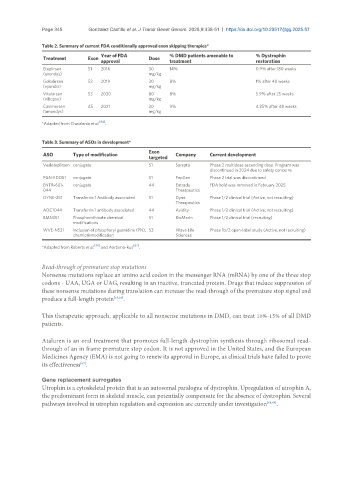
Table 2. Summary of current FDA conditionally approved exon skipping therapies*
Year of FDA % DMD patients amenable to % Dystrophin
Treatment Exon Dose
approval treatment restoration
Eteplirsen 51 2016 30 14% 0.9% after 180 weeks
(exondys) mg/kg
Golodirsen 53 2019 30 8% 1% after 48 weeks
(vyondis) mg/kg
Vitolarsen 53 2020 80 8% 5.9% after 25 weeks
(viltepso) mg/kg
Casimersen 45 2021 30 9% 4.25% after 48 weeks
(amondys) mg/kg
[64]
*Adapted from Chwalenia et al. .
Table 3. Summary of ASOs in development*
Exon
ASO Type of modification Company Current development
targeted
Vesleteplirsen conjugate 51 Sarepta Phase 2 multidose ascending dose. Program was
discontinued in 2024 due to safety concerns
PGN-EDO51 conjugate 51 PepGen Phase 2 trial was discontinued
ENTR-601- conjugate 44 Entrada FDA hold was removed in February 2025
044 Therapeutics
DYNE-251 Transferrin 1 Antibody associated 51 Dyne Phase 1/2 clinical trial (Active, not recruiting)
Therapeutics
AOC1044 Transferrin 1 antibody associated 44 Avidity Phase 1/2 clinical trial (Active, not recruiting)
BMN351 Phosphorothioate chemical 51 BioMarin Phase 1/2 clinical trial (recruiting)
modifications
WVE-N531 Inclusion of phosphoryl guanidine (PN), 53 Wave Life Phase 1b/2 open-label study (Active, not recruiting)
chemical modification Sciences
*Adapted from Roberts et al. [36] and Aartsma-Rus [62] .
Read-through of premature stop mutations
Nonsense mutations replace an amino acid codon in the messenger RNA (mRNA) by one of the three stop
codons - UAA, UGA or UAG, resulting in an inactive, truncated protein. Drugs that induce suppression of
these nonsense mutations during translation can increase the read-through of the premature stop signal and
produce a full-length protein [65,66] .
This therapeutic approach, applicable to all nonsense mutations in DMD, can treat 10%-15% of all DMD
patients.
Ataluren is an oral treatment that promotes full-length dystrophin synthesis through ribosomal read-
through of an in frame premature stop codon. It is not approved in the United States, and the European
Medicines Agency (EMA) is not going to renew its approval in Europe, as clinical trials have failed to prove
its effectiveness .
[67]
Gene replacement surrogates
Utrophin is a cytoskeletal protein that is an autosomal paralogue of dystrophin. Upregulation of utrophin A,
the predominant form in skeletal muscle, can potentially compensate for the absence of dystrophin. Several
pathways involved in utrophin regulation and expression are currently under investigation [68,69] .