
Page 118 - Read Online
P. 118
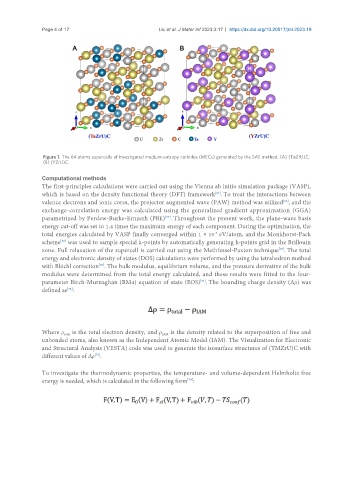
Page 4 of 17 Liu et al. J Mater Inf 2023;3:17 https://dx.doi.org/10.20517/jmi.2023.19
Figure 1. The 64 atoms supercells of investigated medium-entropy carbides (MECs) generated by the SAE method. (A) (TaZrU)C;
(B) (YZrU)C.
Computational methods
The first-principles calculations were carried out using the Vienna ab initio simulation package (VASP),
[65]
which is based on the density functional theory (DFT) framework . To treat the interactions between
valence electrons and ionic cores, the projector augmented wave (PAW) method was utilized , and the
[66]
exchange-correlation energy was calculated using the generalized gradient approximation (GGA)
[67]
parametrized by Perdew-Burke-Ernzerh (PBE) . Throughout the present work, the plane-wave basis
energy cut-off was set to 1.4 times the maximum energy of each component. During the optimization, the
total energies calculated by VASP finally converged within 1 × 10 eV/atom, and the Monkhorst-Pack
-6
scheme was used to sample special k-points by automatically generating k-points grid in the Brillouin
[68]
[69]
zone. Full relaxation of the supercell is carried out using the Methfessel-Paxton technique . The total
energy and electronic density of states (DOS) calculations were performed by using the tetrahedron method
[70]
with Blöchl correction . The bulk modulus, equilibrium volume, and the pressure derivative of the bulk
modulus were determined from the total energy calculated, and these results were fitted to the four-
parameter Birch-Murnaghan (BM4) equation of state (EOS) . The bounding charge density (∆ρ) was
[71]
[72]
defined as :
Where ρ is the total electron density, and ρ is the density related to the superposition of free and
total
IAM
unbonded atoms, also known as the Independent Atomic Model (IAM). The Visualization for Electronic
and Structural Analysis (VESTA) code was used to generate the isosurface structures of (TMZrU)C with
different values of ∆ρ .
[73]
To investigate the thermodynamic properties, the temperature- and volume-dependent Helmholtz free
[74]
energy is needed, which is calculated in the following form :