
Page 185 - Read Online
P. 185
Seo et al. Energy Mater. 2025, 5, 500123 https://dx.doi.org/10.20517/energymater.2025.38 Page 5 of 18
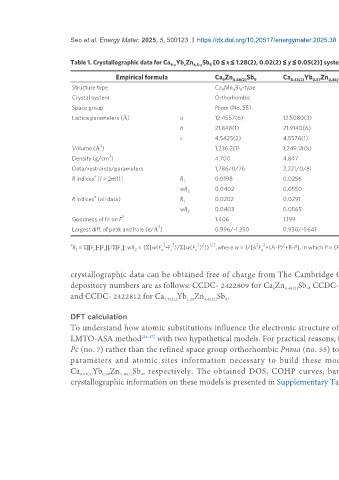
Table 1. Crystallographic data for Ca Yb Zn 4.5-y Sb [0 ≤ x ≤ 1.28(2), 0.02(2) ≤ y ≤ 0.05(2)] system
9-x
x
9
Empirical formula Ca Zn 4.48(2) Sb 9 Ca 8.43(2) Yb 0.57 Zn 4.46(1) Sb 9 Ca 8.01(3) Yb 0.99 Zn 4.46(1) Sb 9 Ca 7.72(2) Yb 1.28 Zn 4.45(2) Sb 9
9
Structure type Ca Mn Bi -type
9 4 9
Crystal system Orthorhombic
Space group Pbam (No. 55)
Lattice parameters (Å) a 12.4557(6) 12.5080(3) 12.4546(4) 12.4523(7)
b 21.848(1) 21.9140(6) 21.8344(6) 21.845(2)
c 4.5425(2) 4.5574(1) 4.5412(1) 4.5438(2)
3
Volume (Å ) 1,236.2(1) 1,249.18(5) 1,234.93(6) 1,236.0(2)
3
Density (g/cm ) 4.700 4.847 5.058 5.151
Data/restraints/parameters 1,786/0/76 2,221/0/81 1,286/0/81 1,947/0/81
a
R indices [I > 2σ(I)] R 0.0198 0.0256 0.0194 0.0176
1
wR 0.0402 0.0550 0.0383 0.0349
2
a
R indices (all data) R 1 0.0202 0.0291 0.0206 0.0223
wR 0.0403 0.0565 0.0386 0.0364
2
2
Goodness of fit on F 1.406 1.199 1.264 1.065
3
Largest diff. of peak and hole (e/Å ) 0.996/-1.350 0.930/-1.641 0.873/-1.329 0.883/-1.017
a 2 2 2 2 1/2 2 2 2 2 2
R = Σ||F |-|F ||/Σ|F |; wR = {Σ[w(F -F )/Σ[w(F ) ]} , where w = 1/[σ F +(A-P) +B-P], in which P = (F + 2F )/3 and A and B are weight coefficients.
1 o c o 2 o c o o o c
crystallographic data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. The
depository numbers are as follows: CCDC- 2422809 for Ca Zn 4.48(2) Sb , CCDC-2422810 for Ca 8.43(2) Yb Zn 4.46(1) Sb , CCDC- 2422811 for Ca 8.01(3) Yb Zn 4.46(1) Sb ,
9
9
0.57
9
0.99
9
and CCDC- 2422812 for Ca 7.72(2) Yb Zn 4.45(2) Sb .
9
1.28
DFT calculation
To understand how atomic substitutions influence the electronic structure of the studied system, DFT calculations were carried out by employing the TB-
LMTO-ASA method [24-27] with two hypothetical models. For practical reasons, these two hypothetical models were designed to have the monoclinic subgroup
Pc (no. 7) rather than the refined space group orthorhombic Pnma (no. 55) to apply the idealized compositions of Ca Zn Sb and Ca YbZn Cu Sb . Lattice
8
9
4
9
0.5
9
4.5
parameters and atomic sites information necessary to build these models were taken from the SXRD refinement results of Ca Zn 4.48(2) Sb and
9
9
Ca 8.01(3) Yb Zn 4.46(1) Sb , respectively. The obtained DOS, COHP curves, band structures, and ELF illustrations were carefully investigated. Detailed
0.99
9
crystallographic information on these models is presented in Supplementary Table 2.