
Page 92 - Read Online
P. 92
Page 4 of 34 West et al. Rare Dis Orphan Drugs J 2024;3:22 https://dx.doi.org/10.20517/rdodj.2023.61
[11]
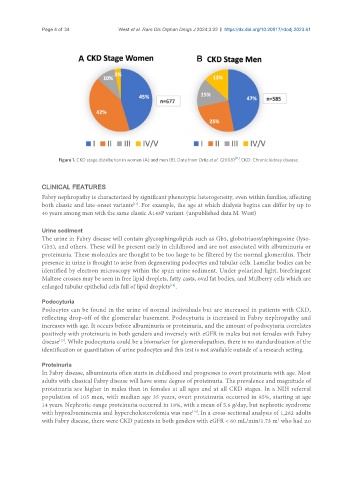
Figure 1. CKD stage distribution in women (A) and men (B). Data from Ortiz et al. (2008) . CKD: Chronic kidney disease.
CLINICAL FEATURES
Fabry nephropathy is characterized by significant phenotypic heterogeneity, even within families, affecting
[17]
both classic and late-onset variants . For example, the age at which dialysis begins can differ by up to
40 years among men with the same classic A143P variant. (unpublished data M. West)
Urine sediment
The urine in Fabry disease will contain glycosphingolipids such as Gb3, globotriaosylsphingosine (lyso-
Gb3), and others. These will be present early in childhood and are not associated with albuminuria or
proteinuria. These molecules are thought to be too large to be filtered by the normal glomerulus. Their
presence in urine is thought to arise from degenerating podocytes and tubular cells. Lamellar bodies can be
identified by electron microscopy within the spun urine sediment. Under polarized light, birefringent
Maltese crosses may be seen in free lipid droplets, fatty casts, oval fat bodies, and Mulberry cells which are
enlarged tubular epithelial cells full of lipid droplets .
[18]
Podocyturia
Podocytes can be found in the urine of normal individuals but are increased in patients with CKD,
reflecting drop-off of the glomerular basement. Podocyturia is increased in Fabry nephropathy and
increases with age. It occurs before albuminuria or proteinuria, and the amount of podocyturia correlates
positively with proteinuria in both genders and inversely with eGFR in males but not females with Fabry
disease . While podocyturia could be a biomarker for glomerulopathies, there is no standardization of the
[19]
identification or quantitation of urine podocytes and this test is not available outside of a research setting.
Proteinuria
In Fabry disease, albuminuria often starts in childhood and progresses to overt proteinuria with age. Most
adults with classical Fabry disease will have some degree of proteinuria. The prevalence and magnitude of
proteinuria are higher in males than in females at all ages and at all CKD stages. In a NIH referral
population of 105 men, with median age 35 years, overt proteinuria occurred in 85%, starting at age
14 years. Nephrotic-range proteinuria occurred in 18%, with a mean of 5.6 g/day, but nephrotic syndrome
with hypoalbuminemia and hypercholesterolemia was rare . In a cross-sectional analysis of 1,262 adults
[10]
2
with Fabry disease, there were CKD patients in both genders with eGFR < 60 mL/min/1.73 m who had no