
Page 134 - Read Online
P. 134
Santiago et al. J Transl Genet Genom 2021;5:380-95 https://dx.doi.org/10.20517/jtgg.2021.16 Page 386
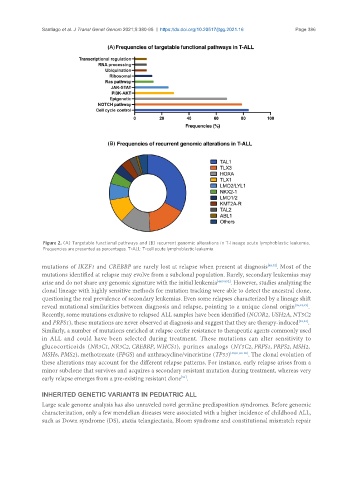
Figure 2. (A) Targetable functional pathways and (B) recurrent genomic alterations in T-lineage acute lymphoblastic leukemia.
Frequencies are presented as percentages. T-ALL: T-cell acute lymphoblastic leukemia.
mutations of IKZF1 and CREBBP are rarely lost at relapse when present at diagnosis [48,52] . Most of the
mutations identified at relapse may evolve from a subclonal population. Rarely, secondary leukemias may
arise and do not share any genomic signature with the initial leukemia [49,50,52] . However, studies analyzing the
clonal lineage with highly sensitive methods for mutation tracking were able to detect the ancestral clone,
questioning the real prevalence of secondary leukemias. Even some relapses characterized by a lineage shift
reveal mutational similarities between diagnosis and relapse, pointing to a unique clonal origin [35,48,49] .
Recently, some mutations exclusive to relapsed ALL samples have been identified (NCOR2, USH2A, NT5C2
and PRPS1), these mutations are never observed at diagnosis and suggest that they are therapy-induced [48,53] .
Similarly, a number of mutations enriched at relapse confer resistance to therapeutic agents commonly used
in ALL and could have been selected during treatment. These mutations can alter sensitivity to
glucocorticoids (NR3C1, NR3C2, CREBBP, WHCS1), purines analogs (NT5C2, PRPS1, PRPS2, MSH2,
MSH6, PMS2), methotrexate (FPGS) and anthracycline/vincristine (TP53) [48,51,53-55] . The clonal evolution of
these alterations may account for the different relapse patterns. For instance, early relapse arises from a
minor subclone that survives and acquires a secondary resistant mutation during treatment, whereas very
early relapse emerges from a pre-existing resistant clone .
[51]
INHERITED GENETIC VARIANTS IN PEDIATRIC ALL
Large scale genome analysis has also unraveled novel germline predisposition syndromes. Before genomic
characterization, only a few mendelian diseases were associated with a higher incidence of childhood ALL,
such as Down syndrome (DS), ataxia telangiectasia, Bloom syndrome and constitutional mismatch repair