
Page 158 - Read Online
P. 158
Page 386 Saneto. J Transl Genet Genom 2020;4:384-428 I http://dx.doi.org/10.20517/jtgg.2020.40
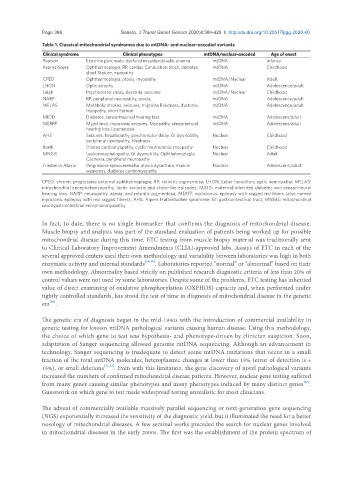
Table 1. Classical mitochondrial syndromes due to mtDNA- and nuclear-encoded variants
Clinical syndrome Clinical phenotypes mtDNA/nuclear-encoded Age of onset
Pearson Exocrine pancreatic dysfunction,sideroblastic anemia mtDNA Infancy
Kearns-Sayre Ophthalmoplegia, RP, cardiac Conduction block, diabetes, mtDNA Childhood
short Stature, myopathy
CPEO Ophthalmoplegia, ptosis, myopathy mtDNA/Nuclear Adult
LHON Optic atrophy mtDNA Adolescence/adult
Leigh Psychomotor delay, dystonia, seizures mtDNA/Nuclear Childhood
NARP RP, peripheral neuropathy, ataxia, mtDNA Adolescence/adult
MELAS Metabolic strokes, seizures, migraine Blindness, dystonia, mtDNA Adolescence/adult
myopathy, short Stature
MIDD Diabetes, sensorineuroal hearing loss mtDNA Adolescent/adult
MERRF Myoclonus, myoclonic seizures, Myopathy, sensorineural mtDNA Adolescent/adult
hearing loss Lipomatosis
AHS Seizures, hepatopathy, psychomotor delay, GI dysmotility, Nuclear Childhood
peripheral neuropathy, blindness
Barth Dilated cardiomyopathy, cyclic neutropenia, myopathy Nuclear Childhood
MNGIE Leukoencephalopathy, GI dysmotility, Ophthalmoplegia, Nuclear Adult
Cachexia, peripheral neuropathy
Friedreich Ataxia Progressive spinocerebellar ataxia dysarthria, muscle Nuclear Adolescent/adult
weakness, diabetes cardiomyopathy
CPEO: chronic progressive external ophthalmoplegia; RP: retinitis pigmentosa; LHON: Leber hereditary optic neuropathy; MELAS:
mitochondrial encephalomyopathy, lactic acidosis and stoke-like episodes; MIDD: maternal-inherited diabetes and sensorineural
hearing loss; NARP: neuropathy, ataxia, and retinitis pigmentosa; MERFF: myoclonus, epilepsy with ragged red fibers (also named
myoclonic epilepsy with red ragged fibers); AHS: Alpers Huttenlocher syndrome; GI: gastrointestinal tract; MNGIE: mitochondrial
neurogastrointestinal encephalomyopathy
In fact, to date, there is no single biomarker that confirms the diagnosis of mitochondrial disease.
Muscle biopsy and analysis was part of the standard evaluation of patients being worked up for possible
mitochondrial disease during this time. ETC testing from muscle biopsy material was traditionally sent
to Clinical Laboratory Improvement Amendments (CLIA)-approved labs. Assays of ETC in each of the
several approved centers used their own methodology and variability between laboratories was high in both
enzymatic activity and internal standards [18,19] . Laboratories reported “normal” or “abnormal” based on their
own methodology. Abnormality based strictly on published research diagnostic criteria of less than 20% of
control values were not used by some laboratories. Despite some of the problems, ETC testing has inherited
value of direct examining of oxidative phosphorylation (OXPHOS) capacity and, when performed under
tightly controlled standards, has stood the test of time in diagnosis of mitochondrial disease in the genetic
[20]
era .
The genetic era of diagnosis began in the mid-1990s with the introduction of commercial availability in
genetic testing for known mtDNA pathological variants causing human disease. Using this methodology,
the choice of which gene to test was hypothesis- and phenotype-driven by clinician suspicion. Soon,
adaptation of Sanger sequencing allowed genome mtDNA sequencing. Although an advancement in
technology, Sanger sequencing is inadequate to detect some mtDNA mutations that occur in a small
fraction of the total mtDNA molecules, heteroplasmic changes at lower than 15% (error of detection is ±
15%), or small deletions [21,22] . Even with this limitation, the gene discovery of novel pathological variants
increased the numbers of confirmed mitochondrial disease patients. However, nuclear gene testing suffered
[23]
from many genes causing similar phenotypes and many phenotypes induced by many distinct genes .
Guesswork on which gene to test made widespread testing unrealistic for most clinicians.
The advent of commercially available massively parallel sequencing or next-generation gene sequencing
(NGS) exponentially increased the sensitivity of the diagnostic yield, but it illuminated the need for a better
nosology of mitochondrial diseases. A few seminal works preceded the search for nuclear genes involved
in mitochondrial diseases in the early 2000s. The first was the establishment of the protein spectrum of