
Page 139 - Read Online
P. 139
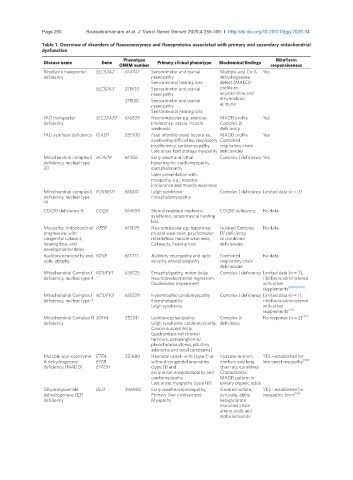
Page 290 Balasubramaniam et al. J Transl Genet Genom 2020;4:285-306 I http://dx.doi.org/10.20517/jtgg.2020.34
Table 1. Overview of disorders of flavocoenzymes and flavoproteins associated with primary and secondary mitochondrial
dysfunction
Disease name Gene Phenotype Primary clinical phenotype Biochemical findings Riboflavin
OMIM number responsiveness
Riboflavin transporter SLC52A2 614707 Sensorimotor and cranial Multiple acyl Co-A Yes
deficiency neuropathy dehydrogenase
Sensorineural hearing loss defect (MADD)
SLC52A3 211500 Sensorimotor and cranial profile on
neuropathy acylcarnitine and
211530 Sensorimotor and cranial ethylmalonic
neuropathy aciduria
Sensorineural hearing loss
FAD transporter SLC25A32 616839 Neuromuscular e.g. exercise MADD profile Yes
deficiency intolerance, ataxia, muscle Complex II
weakness deficiency
FAD synthase deficiency FLAD1 255100 Fatal infantile onset hypotonia, MADD profile Yes
swallowing difficulties, respiratory Combined
insufficiency, cardiomyopathy respiratory chain
Late onset lipid storage myopathy deficiencies
Mitochondrial complex I ACAD9 611126 Early onset and lethal Complex I deficiency Yes
deficiency, nuclear type hypertrophic cardiomyopathy,
20 encephalopathy
Later presentation with
myopathy, e.g., exercise
intolerance and muscle weakness
Mitochondrial complex I FOXRED1 618241 Leigh syndrome Complex I deficiency Limited data (n = 2)
deficiency, nuclear type Encephalomyopathy
19
COQ10 deficiency-6 COQ6 614650 Steroid resistant nephrotic COQ10 deficiency No data
syndrome, sensorineural hearing
loss
Myopathy, mitochondrial GFER 613076 Neuromuscular e.g. hypotonia, Isolated Complex No data
progressive, with muscle weakness, psychomotor IV deficiency
congenital cataract, retardation, muscle weakness, or combined
hearing loss, and Cataracts, hearing loss deficiencies
developmental delay
Auditory neuropathy and FDXR 617717 Auditory neuropathy and optic Combined No data
optic atrophy atrophy, encephalopathy respiratory chain
deficiencies
Mitochondrial Complex I NDUFV1 618225 Encephalopathy, motor delay Complex I deficiency Limited data (n = 7),
deficiency, nuclear type 4 Neurodevelopmental regression, riboflavin administered
Oculomotor impairment with other
supplements [94,97,98,101]
Mitochondrial Complex I NDUFV2 618229 Hypertrophic cardiomyopathy Complex I deficiency Limited data (n = 1),
deficiency, nuclear type 7 Encephalopathy riboflavin administered
Leigh syndrome with other
supplements [113]
Mitochondrial Complex II SDHA 252011 Leukoencephalopathy, Complex II No response (n = 2) [133]
deficiency Leigh syndrome, cardiomyopathy, deficiency
Cancer susceptibility
(gastrointestinal stromal
tumours, paraganglioma/
pheochromocytoma, pituitary
adenoma and renal carcinoma)
Multiple acyl-coenzyme ETFA, 231680 Neonatal onset- with (type I) or Increase in short, YES - established for
A dehydrogenase ETFB, without congenital anomalies medium and long late onset myopathy [138]
deficiency (MADD) ETFDH (type II) and chain acylcarnitines
early onset encephalopathy and Characteristic
cardiomyopathy MADD pattern in
Late onset myopathy (type III) urinary organic acids
Dihydrolipoamide DLD 246900 Early onset encephalopathy, Elevated lactate, YES - established for
dehydrogenase (E3) Primary liver involvement, pyruvate, alpha myopathic form [146]
deficiency Myopathy ketoglutarate,
branched chain
amino acids and
alpha ketoacids