
Page 96 - Read Online
P. 96
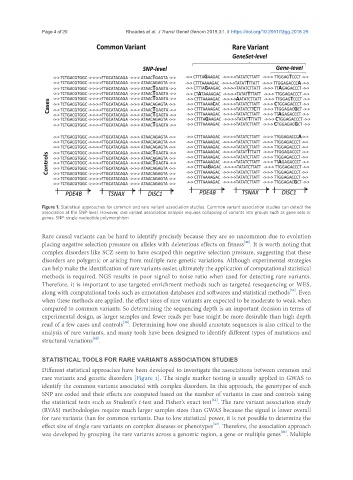
Page 4 of 20 Rhoades et al. J Transl Genet Genom 2019;3:1. I https://doi.org/10.20517/jtgg.2018.26
Figure 1. Statistical approaches for common and rare variant association studies. Common variant association studies can detect the
association at the SNP-level. However, rare variant association analysis requires collapsing of variants into groups such as gene sets or
genes. SNP: single nucleotide polymorphism
Rare causal variants can be hard to identify precisely because they are so uncommon due to evolution
[40]
placing negative selection pressure on alleles with deleterious effects on fitness . It is worth noting that
complex disorders like SCZ seem to have escaped this negative selection pressure, suggesting that these
disorders are polygenic or arising from multiple rare genetic variations. Although experimental strategies
can help make the identification of rare variants easier, ultimately the application of computational statistical
methods is required. NGS results in poor signal to noise ratio when used for detecting rare variants.
Therefore, it is important to use targeted enrichment methods such as targeted resequencing or WES,
[38]
along with computational tools such as annotation databases and softwares and statistical methods . Even
when these methods are applied, the effect sizes of rare variants are expected to be moderate to weak when
compared to common variants. So determining the sequencing depth is an important decision in terms of
experimental design, as larger samples and fewer reads per base might be more desirable than high depth
[40]
read of a few cases and controls . Determining how one should annotate sequences is also critical to the
analysis of rare variants, and many tools have been designed to identify different types of mutations and
[41]
structural variations .
sTaTIsTICal TOOls fOR RaRe VaRIaNTs assOCIaTION sTUDIes
Different statistical approaches have been developed to investigate the associations between common and
rare variants and genetic disorders [Figure 1]. The single marker testing is usually applied in GWAS to
identify the common variants associated with complex disorders. In this approach, the genotypes of each
SNP are coded and their effects are computed based on the number of variants in case and controls using
[42]
the statistical tests such as Student’s t-test and Fisher’s exact test . The rare variant association study
(RVAS) methodologies require much larger samples sizes than GWAS because the signal is lower overall
for rare variants than for common variants. Due to low statistical power, it is not possible to determine the
[43]
effect size of single rare variants on complex diseases or phenotypes . Therefore, the association approach
[40]
was developed by grouping the rare variants across a genomic region, a gene or multiple genes . Multiple