
Page 22 - Read Online
P. 22
Page 6 of 9 Hong et al. J Transl Genet Genom 2018;2:8. I https://doi.org/10.20517/jtgg.2018.06
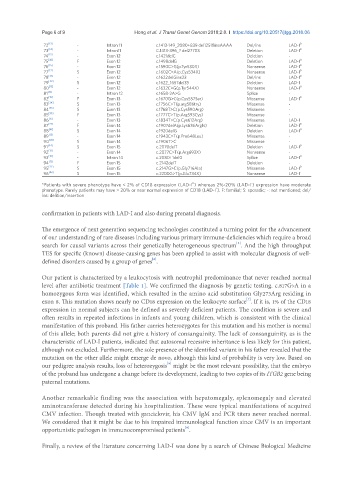
72 [13] - Intron 11 c.1413-149_2080+839 del12518insAAAA Del/ins LAD-I 0
73 [14] - Intron11 C.1413-396_? del27703 Deletion LAD-I 0
74 [13] - Exon 12 c.1421delC Deletion -
75 [41] F Exon 12 c.1498delG Deletion LAD-I 0
76 [15] - Exon 12 c.1590C>G(p.Tyr530X) Nonsense LAD-I 0
77 [17] S Exon 12 c.1602C>A(p.Cys534X) Nonsense LAD-I 0
78 [13] - Exon 12 c.1622delGins23 Del/ins LAD-I 0
79 [42] S Exon 12 c.1622_1657del33 Deletion LAD-I -
80 [13] - Exon 12 c.1632C>G(p.Tyr544X) Nonsense LAD-I 0
81 [13] - Intron 12 c.1658-2A>G Splice -
82 [15] F Exon 13 c.1670G>C(p.Cys557Ser) Missense LAD-I 0
83 [34] S Exon 13 c.1756C>T(p.arg586try) Missense -
84 [35] S Exon 13 c.1768T>C(p.Cys590Arg) Missense -
85 [35] F Exon 13 c.1777C>T(p.Arg593Cys) Missense -
86 [15] - Exon 13 c.1834T>C(p.Cys612Arg) Missense LAD-I -
87 [22] F Exon 14 c.1907delA(p.Lys636Argfs) Deletion LAD-I 0
88 [41] S Exon 14 c.1920delG Deletion LAD-I 0
89 [13] - Exon 14 c.1943C>T(p.Pro648Leu) Missense -
90 [43] S Exon 14 c.1906T>C Missense -
91 [44] S Exon 15 c.2070delT Deletion LAD-I 0
92 [13] - Exon 14 c.2077C>T(p.Arg693X) Nonsense -
93 [13] - Intron 14 c.2080+1delG Splice LAD-I 0
94 [12] F Exon 15 c.2142delT Deletion -
95 [22] S Exon 15 c.2147G>C(p.Gly716Ala) Missense LAD-I 0
96 [42] S Exon 15 c.2200G>T(p.Glu734X) Nonsense LAD-I -
-
0
*Patients with severe phenotype have < 2% of CD18 expression (LAD-I ) whereas 2%-20% (LAD-I ) expression have moderate
+
phenotype. Rarely patients may have > 20% or near normal expression of CD18 (LAD-I ). F: familial; S: sporadic; -: not mentioned; del/
ins: delition/insertion
confirmation in patients with LAD-I and also during prenatal diagnosis.
The emergence of next generation sequencing technologies constituted a turning point for the advancement
of our understanding of rare diseases including various primary immune-deficiencies which require a broad
search for causal variants across their genetically heterogeneous spectrum . And the high throughput
[5]
TES for specific (known) disease-causing genes has been applied to assist with molecular diagnosis of well-
defined disorders caused by a group of genes .
[6]
Our patient is characterized by a leukocytosis with neutrophil predominance that never reached normal
level after antibiotic treatment [Table 1]. We confirmed the diagnosis by genetic testing. c.817G>A in a
homozygous form was identified, which resulted in the amino acid substitution Gly273Arg residing in
[7]
exon 8. This mutation shows nearly no CD18 expression on the leukocyte surface . If it is, 1% of the CD18
expression in normal subjects can be defined as severely deficient patients. The condition is severe and
often results in repeated infections in infants and young children, which is consistent with the clinical
manifestation of this proband. His father carries heterozygotes for this mutation and his mother is normal
of this allele; both parents did not give a history of consanguinity. The lack of consanguinity, as is the
characteristic of LAD-I patients, indicated that autosomal recessive inheritance is less likely for this patient,
although not excluded. Furthermore, the sole presence of the identified variant in his father revealed that the
mutation on the other allele might emerge de novo, although this kind of probability is very low. Based on
[8]
our pedigree analysis results, loss of heterozygosis might be the most relevant possibility, that the embryo
of the proband has undergone a change before its development, leading to two copies of its ITGB2 gene being
paternal mutations.
Another remarkable finding was the association with hepatomegaly, splenomegaly and elevated
aminotransferase detected during his hospitalization. These were typical manifestations of acquired
CMV infection. Though treated with ganciclovir, his CMV IgM and PCR titers never reached normal.
We considered that it might be due to his impaired immunological function since CMV is an important
[9]
opportunistic pathogen in immunocompromised patients .
Finally, a review of the literature concerning LAD-I was done by a search of Chinese Biological Medicine