
Page 87 - Read Online
P. 87
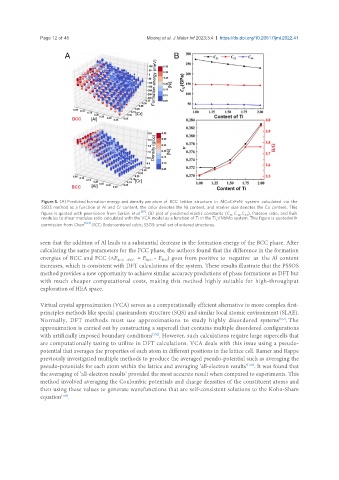
Page 12 of 45 Mooraj et al. J Mater Inf 2023;3:4 https://dx.doi.org/10.20517/jmi.2022.41
Figure 5. (A) Predicted formation energy and density per atom of BCC lattice structure in AlCoCrFeNi system calculated via the
SSOS method as a function of Al and Cr content, the color denotes the Ni content, and marker size denotes the Co content. This
figure is quoted with permission from Sorkin et al. [92] ; (B) plot of predicted elastic constants (C , C , C ), Poisson ratio, and Bulk
12
44
11
modulus to shear modulus ratio calculated with the VCA model as a function of Ti in the Ti VNbMo system. This figure is quotedwith
x
permission from Chen [104] . BCC: Body-centered cubic; SSOS: small set of ordered structures.
seen that the addition of Al leads to a substantial decrease in the formation energy of the BCC phase. After
calculating the same parameters for the FCC phase, the authors found that the difference in the formation
energies of BCC and FCC (ΔE BCC→FCC = E - E ) goes from positive to negative as the Al content
FCC
BCC
increases, which is consistent with DFT calculations of the system. These results illustrate that the PSSOS
method provides a new opportunity to achieve similar accuracy predictions of phase formations as DFT but
with much cheaper computational costs, making this method highly suitable for high-throughput
exploration of HEA space.
Virtual crystal approximation (VCA) serves as a computationally efficient alternative to more complex first-
principles methods like special quasirandom structure (SQS) and similar local atomic environment (SLAE).
Normally, DFT methods must use approximations to study highly disordered systems . The
[102]
approximation is carried out by constructing a supercell that contains multiple disordered configurations
with artificially imposed boundary conditions . However, such calculations require large supercells that
[102]
are computationally taxing to utilize in DFT calculations. VCA deals with this issue using a pseudo-
potential that averages the properties of each atom in different positions in the lattice cell. Ramer and Rappe
previously investigated multiple methods to produce the averaged pseudo-potential such as averaging the
pseudo-potentials for each atom within the lattice and averaging ‘all-electron results’ . It was found that
[103]
the averaging of ‘all-electron results’ provided the most accurate result when compared to experiments. This
method involved averaging the Coulombic potentials and charge densities of the constituent atoms and
then using these values to generate wavefunctions that are self-consistent solutions to the Kohn-Sham
equation .
[103]