
Page 113 - Read Online
P. 113
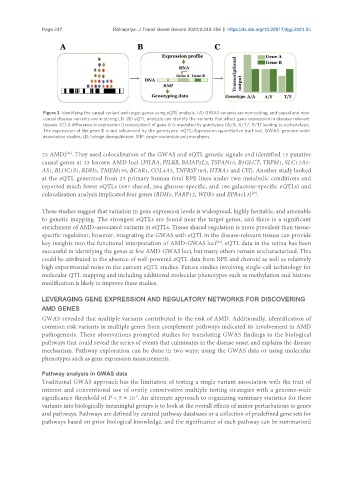
Page 247 Ratnapriya. J Transl Genet Genom 2022;6:240-256 https://dx.doi.org/10.20517/jtgg.2021.54
Figure 3. Identifying the causal variant and target genes using eQTL analysis. (A) GWAS variants are non-coding, and causal and non-
causal disease variants are in strong LD. (B) eQTL analysis can identify the variants that affect gene expression in disease-relevant
tissues. (C) A difference in expression (transcription) of gene A is mediated by genotypes (A/A, A/T/, T/T) leading to a phenotype.
The expression of the gene B is not influenced by the genotypes. eQTL: Expression quantitative trait loci; GWAS: genome-wide
association studies; LD: linkage disequilibrium; SNP: single nucleotide polymorphism.
23 AMD) . They used colocalization of the GWAS and eQTL genetic signals and identified 15 putative
[86]
causal genes at 13 known AMD loci (PILRA, PILRB, BAIAP2L2, TSPAN10, B3GLCT, TRPM1, SLC12A5-
AS1, BLOC1S1, RDH5, TMEM199, BCAR1, COL4A3, TNFRSF10A, HTRA1 and CFI). Another study looked
at the eQTL generated from 23 primary human fetal RPE lines under two metabolic conditions and
reported much fewer eQTLs (687 shared, 264 glucose-specific, and 166 galactose-specific eQTLs) and
colocalization analysis implicated four genes (RDH5, PARP12, WDR5 and EPB41L3) .
[87]
These studies suggest that variation in gene expression levels is widespread, highly heritable, and amenable
to genetic mapping. The strongest eQTLs are found near the target genes, and there is a significant
enrichment of AMD-associated variants in eQTLs. Tissue shared regulation is more prevalent than tissue-
specific regulation; however, integrating the GWAS with eQTL in the disease-relevant tissues can provide
[80]
key insights into the functional interpretation of AMD-GWAS loci . eQTL data in the retina has been
successful in identifying the genes at few AMD-GWAS loci, but many others remain uncharacterized. This
could be attributed to the absence of well-powered eQTL data from RPE and choroid as well as relatively
high experimental noise in the current eQTL studies. Future studies involving single-cell technology for
molecular QTL mapping and including additional molecular phenotypes such as methylation and histone
modification is likely to improve these studies.
LEVERAGING GENE EXPRESSION AND REGULATORY NETWORKS FOR DISCOVERING
AMD GENES
GWAS revealed that multiple variants contributed to the risk of AMD. Additionally, identification of
common risk variants in multiple genes from complement pathways indicated its involvement in AMD
pathogenesis. These observations prompted studies for translating GWAS findings to the biological
pathways that could reveal the series of events that culminates in the disease onset and explains the disease
mechanism. Pathway exploration can be done in two ways: using the GWAS data or using molecular
phenotypes such as gene expression measurements.
Pathway analysis in GWAS data
Traditional GWAS approach has the limitation of testing a single variant association with the trait of
interest and conventional use of overly conservative multiple testing strategies with a genome-wide
-8
significance threshold of P < 5 × 10 . An alternate approach to organizing summary statistics for these
variants into biologically meaningful groups is to look at the overall effects of minor perturbations to genes
and pathways. Pathways are defined by curated pathway databases or a collection of predefined gene sets for
pathways based on prior biological knowledge, and the significance of each pathway can be summarized