
Page 31 - Read Online
P. 31
Oquendo et al. J Transl Genet Genom 2021;5:89-111 https://dx.doi.org/10.20517/jtgg.2021.04 Page 103
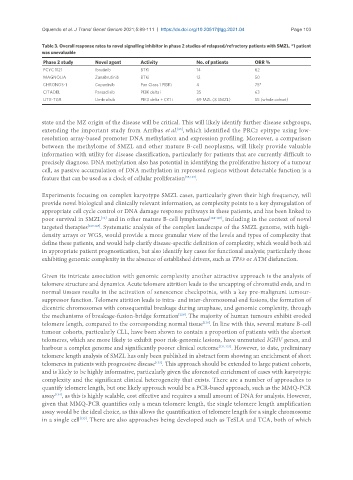
Table 3. Overall response rates to novel signalling inhibitor in phase 2 studies of relapsed/refractory patients with SMZL. *1 patient
was unevaluable
Phase 2 study Novel agent Activity No. of patients ORR %
PCYC 1121 Ibrutinib BTKi 14 62
MAGNOLIA Zanabrutinib BTKi 12 50
CHRONOS-1 Copanlisib Pan Class 1 PI3Ki 4 75*
CITADEL Parsaclisib PI3K delta i 35 63
UTX-TGR Umbralisib PIK3 delta + CK1 i 69 MZL (8 SMZL) 55 (whole cohort)
state and the MZ origin of the disease will be critical. This will likely identify further disease subgroups,
[25]
extending the important study from Arribas et al. , which identified the PRC2 epitype using low-
resolution array-based promoter DNA methylation and expression profiling. Moreover, a comparison
between the methylome of SMZL and other mature B-cell neoplasms, will likely provide valuable
information with utility for disease classification, particularly for patients that are currently difficult to
precisely diagnose. DNA methylation also has potential in identifying the proliferative history of a tumour
cell, as passive accumulation of DNA methylation in repressed regions without detectable function is a
feature that can be used as a clock of cellular proliferation [95,123] .
Experiments focusing on complex karyotype SMZL cases, particularly given their high frequency, will
provide novel biological and clinically relevant information, as complexity points to a key dysregulation of
appropriate cell cycle control or DNA damage response pathways in these patients, and has been linked to
poor survival in SMZL and in other mature B-cell lymphomas [124-126] , including in the context of novel
[31]
targeted therapies [127,128] . Systematic analysis of the complex landscape of the SMZL genome, with high-
density arrays or WGS, would provide a more granular view of the levels and types of complexity that
define these patients, and would help clarify disease-specific definition of complexity, which would both aid
in appropriate patient prognostication, but also identify key cases for functional analysis; particularly those
exhibiting genomic complexity in the absence of established drivers, such as TP53 or ATM disfunction.
Given its intricate association with genomic complexity another attractive approach is the analysis of
telomere structure and dynamics. Acute telomere attrition leads to the uncapping of chromatid ends, and in
normal tissues results in the activation of senescence checkpoints, with a key pre-malignant tumour-
suppressor function. Telomere attrition leads to intra- and inter-chromosomal end fusions, the formation of
dicentric chromosomes with consequential breakage during anaphase, and genomic complexity, through
[129]
the mechanisms of breakage-fusion-bridge formation . The majority of human tumours exhibit eroded
telomere length, compared to the corresponding normal tissue . In line with this, several mature B-cell
[130]
tumour cohorts, particularly CLL, have been shown to contain a proportion of patients with the shortest
telomeres, which are more likely to exhibit poor risk-genomic lesions, have unmutated IGHV genes, and
harbour a complex genome and significantly poorer clinical outcome [131,132] . However, to date, preliminary
telomere length analysis of SMZL has only been published in abstract form showing an enrichment of short
[133]
telomeres in patients with progressive disease . This approach should be extended to large patient cohorts,
and is likely to be highly informative, particularly given the aforenoted enrichment of cases with karyotypic
complexity and the significant clinical heterogeneity that exists. There are a number of approaches to
quantify telomere length, but one likely approach would be a PCR-based approach, such as the MMQ-PCR
assay , as this is highly scalable, cost effective and requires a small amount of DNA for analysis. However,
[134]
given that MMQ-PCR quantifies only a mean telomere length, the single telomere length amplification
assay would be the ideal choice, as this allows the quantification of telomere length for a single chromosome
in a single cell . There are also approaches being developed such as TeSLA and TCA, both of which
[135]