
Page 132 - Read Online
P. 132
Yasin et al. J Transl Genet Genom 2020;4:307-19 I https://doi.org/10.20517/jtgg.2020.30 Page 313
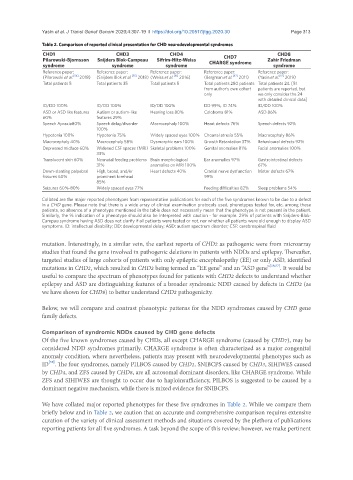
Table 2. Comparison of reported clinical presentation for CHD neurodevelopmental syndromes
CHD1 CHD3 CHD4 CHD8
CHD7
Pilarowski-Bjornsson Snijders Blok-Campeau Sifrim-Hitz-Weiss CHARGE syndrome Zahir Friedman
syndrome syndrome syndrome syndrome
Reference paper: Reference paper: Reference paper: Reference paper: Reference paper:
(Pilarowski et al. [54] 2018) (Snijders Blok et al. [55] 2018) (Weiss et al. [81] 2016) (Bergman et al. [82] 2011) (Yasin et al. [52] 2019)
Total patients 5 Total patients 35 Total patients 5 Total patients 280 patients Total patients 24. [51
from author’s own cohort patients are reported, but
only we only consider the 24
with detailed clinical data]
ID/DD 100% ID/DD 100% ID/DD 100% DD 99%, ID 74% ID/DD 100%
ASD or ASD like features Autism or autism-like Hearing loss 80% Coloboma 81% ASD 86%
60% features 29%
Speech Apraxia80% Speech delay/disorder Macrocephaly 100% Heart defects 76% Speech defects 92%
100%
Hypotonia 100% Hypotonia 75% Widely spaced eyes 100% Choanal atresia 55% Macrocephaly 86%
Macrocephaly 40% Macrocephaly 58% Dysmorphic ears 100% Growth Retardation 37% Behavioural defects 92%
Depressed midface 60% Widened CSF spaces (MRI) Skeletal problems 100% Genital anomalies 81% Facial anomalies 100%
33%
Translucent skin 60% Neonatal feeding problems Brain morphological Ear anomalies 97% Gastrointestinal defects
31% anomalies on MRI 100% 67%
Down-slanting palpebral High, broad, and/or Heart defects 40% Cranial nerve dysfunction Motor defects 67%
fissures 60% prominent forehead 99%
85%
Seizures 60%-80% Widely spaced eyes 77% Feeding difficulties 82% Sleep problems 54%
Collated are the major reported phenotypes from representative publications for each of the five syndromes known to be due to a defect
in a CHD gene. Please note that there is a wide array of clinical examination protocols used, phenotypes tested for, etc. among these
patients, so absence of a phenotype mentioned in the table does not necessarily mean that the phenotype is not present in the patient.
Similarly, the % indication of a phenotype should also be interpreted with caution - for example. 29% of patients with Snijders-Blok-
Campau syndrome having ASD does not clarify if all patients were tested or not, nor whether all patients were old enough to display ASD
symptoms. ID: intellectual disability; DD: developmental delay; ASD: autism spectrum disorder; CSF: cerebrospinal fluid
mutation. Interestingly, in a similar vein, the earliest reports of CHD2 as pathogenic were from microarray
studies that found the gene involved in pathogenic deletions in patients with NDDs and epilepsy. Thereafter,
targeted studies of large cohorts of patients with only epileptic encephalopathy (EE) or only ASD, identified
mutations in CHD2, which resulted in CHD2 being termed an “EE gene” and an “ASD gene” [29,57] . It would be
useful to compare the spectrum of phenotypes found for patients with CHD2 defects to understand whether
epilepsy and ASD are distinguishing features of a broader syndromic NDD caused by defects in CHD2 (as
we have shown for CHD8) to better understand CHD2 pathogenicity.
Below, we will compare and contrast phenotypic patterns for the NDD syndromes caused by CHD gene
family defects.
Comparison of syndromic NDDs caused by CHD gene defects
Of the five known syndromes caused by CHDs, all except CHARGE syndrome (caused by CHD7), may be
considered NDD syndromes primarily. CHARGE syndrome is often characterized as a major congenital
anomaly condition, where nevertheless, patients may present with neurodevelopmental phenotypes such as
ID . The four syndromes, namely PILBOS caused by CHD1, SNIBCPS caused by CHD3, SIHIWES caused
[58]
by CHD4, and ZFS caused by CHD8, are all autosomal dominant disorders, like CHARGE syndrome. While
ZFS and SIHIWES are thought to occur due to haploinsufficiency, PILBOS is suggested to be caused by a
dominant negative mechanism, while there is mixed evidence for SNIBCPS.
We have collated major reported phenotypes for these five syndromes in Table 2. While we compare them
briefly below and in Table 2, we caution that an accurate and comprehensive comparison requires extensive
curation of the variety of clinical assessment methods and situations covered by the plethora of publications
reporting patients for all five syndromes. A task beyond the scope of this review; however, we make pertinent