
Page 39 - Read Online
P. 39
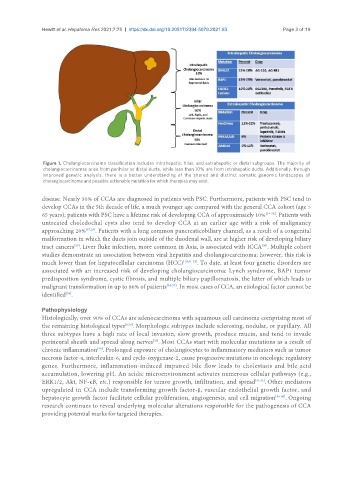
Hewitt et al. Hepatoma Res 2021;7:75 https://dx.doi.org/10.20517/2394-5079.2021.83 Page 3 of 19
Figure 1. Cholangiocarcinoma classification includes intrahepatic, hilar, and extrahepatic or distal subgroups. The majority of
cholangiocarcinomas arise from perihilar or distal ducts, while less than 10% are from intrahepatic ducts. Additionally, through
improved genetic analysis, there is a better understanding of the shared and distinct somatic genomic landscapes of
cholangiocarcinoma and possible actionable mutation for which therapies may exist.
disease. Nearly 30% of CCAs are diagnosed in patients with PSC. Furthermore, patients with PSC tend to
develop CCAs in the 5th decade of life, a much younger age compared with the general CCA cohort (age >
65 years); patients with PSC have a lifetime risk of developing CCA of approximately 10% [24-26] . Patients with
untreated choledochal cysts also tend to develop CCA at an earlier age with a risk of malignancy
approaching 20% [27,28] . Patients with a long common pancreaticobiliary channel, as a result of a congenital
malformation in which the ducts join outside of the duodenal wall, are at higher risk of developing biliary
tract cancers . Liver fluke infection, more common in Asia, is associated with ICCA . Multiple cohort
[29]
[30]
studies demonstrate an association between viral hepatitis and cholangiocarcinoma; however, this risk is
much lower than for hepatocellular carcinoma (HCC) [18,31-33] . To date, at least four genetic disorders are
associated with an increased risk of developing cholangiocarcinoma: Lynch syndrome, BAP1 tumor
predisposition syndrome, cystic fibrosis, and multiple biliary papillomatosis, the latter of which leads to
malignant transformation in up to 80% of patients [34-37] . In most cases of CCA, an etiological factor cannot be
identified .
[38]
Pathophysiology
Histologically, over 90% of CCAs are adenocarcinoma with squamous cell carcinoma comprising most of
the remaining histological types [9,39] . Morphologic subtypes include sclerosing, nodular, or papillary. All
three subtypes have a high rate of local invasion, slow growth, produce mucin, and tend to invade
[39]
perineural sheath and spread along nerves . Most CCAs start with molecular mutations as a result of
chronic inflammation . Prolonged exposure of cholangiocytes to inflammatory mediators such as tumor
[40]
necrosis factor-α, interleukin-6, and cyclo-oxygenase-2, cause progressive mutations in oncologic regulatory
genes. Furthermore, inflammation-induced impaired bile flow leads to cholestasis and bile acid
accumulation, lowering pH. An acidic microenvironment activates numerous cellular pathways (e.g.,
ERK1/2, Akt, NF-κB, etc.) responsible for tumor growth, infiltration, and spread [41,42] . Other mediators
upregulated in CCA include transforming growth factor-β, vascular endothelial growth factor, and
hepatocyte growth factor facilitate cellular proliferation, angiogenesis, and cell migration [43-46] . Ongoing
research continues to reveal underlying molecular alterations responsible for the pathogenesis of CCA
providing potential marks for targeted therapies.